
Abstract
Background:
Tumor genomic profiling has a significant impact on the selection of targeted therapy. Circulating tumor DNA (ctDNA) has emerged as a noninvasive, and reproducible assay compared with tissue biopsy. We aimed to evaluate its utility in identifying mutations and guiding targeted therapy for lung cancer.
Methods:
A total of 173 lung cancer patients underwent next-generation sequencing (NGS) using a targeted enrichment panel covering 20 lung cancer-related genes. The performance of the ctDNA NGS assay in identifying genetic mutations or alterations was compared with tissue biopsy and droplet digital PCR (ddPCR). The treatment response to epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) therapies based on the ctDNA assay results was also assessed.
Results:
The ctDNA was detected in 61.85% of patients. Tissue mutations were detected in paired ctDNA in 38.57% of cases, while ctDNA mutations were detected in paired tissues in 89.1% of cases. The ctDNA increased the number of advanced non-small cell lung cancer (NSCLC) patients who received NCCN-recommended genetic testing by 12%. The concordance between ddPCR and ctDNA was relatively high reaching 99.43%. EGFR T790M/C797S c.G2390C and EGFR T790M/C797S c.T2389A were detected in tissue and ctDNA, respectively, in patient 01015. Moreover, ctDNA assay identified the EGFR T790M mutation, which was missed by tissue biopsy in patient 01149, who developed drug resistance after 1 year of EGFR-TKI therapy. Of the 17 patients who received EGFR-TKI targeted therapies based on the ctDNA NGS results, 12 patients achieved a partial response and two patients had stable disease.
Conclusions:
The results demonstrated that the ctDNA assay could partially overcome tumor heterogeneity in detecting mutations and provide complementary information on tumor genomic profiles. Moreover, the presence of EGFR mutations in ctDNA could offer valuable guidance for selecting appropriate EGFR-TKI treatment for advanced lung cancer patients. However, it is important to note that the ctDNA NGS assay has certain limitations in fully identifying all genomic alterations present in the tumor.
Introduction
Lung cancer is the most common malignant tumor of the respiratory system and lung cancer ranked as the most common cancer not only in terms of morbidity but also mortality in China. 1 Despite the remarkable progress in chemotherapy, the 5-year survival rate for lung cancer remains very low, and the overall treatment effect is not satisfactory. 2 Most lung cancer patients are diagnosed at intermediate and advanced stages, and their tumors do not respond well to therapy. Targeted therapy aimed to actionable alterations has improved the management of lung cancer and revolutionized treatment outcomes compared with chemotherapy. 3 The National Comprehensive Cancer Network (NCCN) guidelines recommend targeted therapy as the preferred treatment option when a reliable driver gene mutation is detected. 2 Common lung cancer targeted genes include anaplastic lymphoma kinase (ALK), epidermal growth factor receptor (EGFR), Kirsten rat sarcoma virus oncogene (KRAS), mesenchymal transition factor (MET), tyrosine receptor kinase ROS proto-oncogene 1 (ROS1), serine–threonine protein kinase B-RAF (BRAF), neurotrophic receptor tyrosine kinase 1,2,3 (NTRK1,2,3), kinesin family member 5B-ret proto-oncogene (RET), and EGFR 2 (ERBB2).
Tissue biopsy remains the gold standard for profiling lung cancer genomic characteristics for molecular targeted therapy.4,5 However, the invasive nature of tissue biopsy and the challenges associated with obtaining repeated samples can limit the ability to effectively monitor response to treatment over time. 6 Due to the existence of tumor heterogeneity, genetic testing of one single tissue biopsy cannot accurately reflect the comprehensive genomic characteristics of the tumor. 7 Tumor evolution and treatment selection pressures lead to the emergence of drug resistance genes, and relying solely on pre-treatment specimens to guide subsequent treatment may lead to suboptimal treatment options. 8 Circulating tumor DNA (ctDNA) is tumor-derived fragmented DNA found in the blood and contains mutations from multiple tumor regions, which provide more comprehensive genomic information. The ctDNA is becoming an appealing alternative due to its noninvasive nature and reproducible, addressing the disadvantages of tissue biopsy. The reliability of ctDNA analysis has been validated in numerous studies, demonstrating its accuracy in capturing the molecular characteristics of tumors compared with tissue biopsies.9-14 In a large-scale prospective study, Leighl and colleagues reported a ctDNA sensitivity of 80% for guideline-recommended biomarkers in 228 patients with newly diagnosed metastatic non-small cell lung cancer (NSCLC). The ctDNA assay increased detection by 48% in addition to tissue biopsy. 14 In another genomic profiling study of advanced NSCLC patients, the concordance between tissue and liquid biopsy for EGFR alterations was 94.0%. 10 These studies have consistently shown a high degree of concordance between mutant profiles of tumor tissues and ctDNAs, establishing a robust foundation for the application of ctDNA in lung cancer patients. While ctDNA holds immense potential in the field of lung cancer, additional research and clinical validation are still necessary to ensure its accuracy and reliability in clinical practice. Furthermore, the development of standardized ctDNA analysis procedures and guidelines is crucial to ensure consistency and comparability in its clinical applications.
To further understand the usefulness of ctDNA in identifying mutations and guiding treatment in clinical practice, 173 lung cancer samples were sequenced. The performance of the ctDNA assay was evaluated in comparison to tissue biopsy and droplet digital PCR (ddPCR) was evaluated. In addition, the treatment response to EGFR-targeted therapy based on the ctDNA assay results was also assessed.
Materials and Methods
Patients
A total of 182 patients diagnosed with lung cancer were recruited from Beijing Chest Hospital between October 2020 and May 2021. Detailed clinicopathological characteristics, including age, sex, clinical stage, and pathology, were collected from the medical records of these patients. The TNM staging was determined according to the American Joint Committee on Cancer (AJCC, Eighth Edition) TNM staging system for lung cancer. The inclusion criteria for patient selection were as follows: (1) age 18 years or older, (2) availability of a peripheral blood sample of 14 to 20 mL for collection, (3) voluntary provision of informed consent, and (4) prior receipt of systemic therapy. Patients were excluded according to the following criteria: (1) inadequate nucleic acid quality after DNA extraction, (2) diagnosed with a second primary malignant tumor, (3) history of prior transplant surgery, and (4) receipt of allogeneic blood transfusion or immunotherapy that could introduce foreign DNA. Nine patients were excluded due to receipt of allogeneic blood transfusion within 1 year, second primary tumors, low library yield, or poor sequencing quality. Among the 173 patients, 107 had paired tumor tissue samples. Concordance analysis between tumor tissue NGS and ctDNA NGS was conducted within this subset of 107 patients. Of the 173 patients, 71 had sufficient DNA for digital droplet PCR (ddPCR) testing after the ctDNA NGS test. The concordance between ddPCR and ctDNA NGS results was analyzed in these 71 patients. In addition, the utility of ctDNA NGS in guiding targeted therapy was analyzed in 17 samples, for whom targeted therapy response based on the ctDNA NGS results was available (see Figure 1).

CONSORT flow diagram of the study participants.
Sample processing and DNA extraction
The blood samples were collected using 10 mL cell-free DNA (cfDNA) BCT tubes (Streck, La Vista, NE). Whole-blood samples were stored at ambient temperature and processed within 1 week of collection. A two-step centrifugation protocol was used to separate the samples into plasma and buffy coat. The tissue and plasma samples were stored at –80°C until DNA isolation. Circulating cfDNA was isolated from 4 mL of plasma using the Nucleic Acid Extraction Kit (Beijing USCI Medical Devices Co., Ltd., TQ003, China). Isolated cfDNA was eluted in 52 μL of elution buffer supplied with the kit, and processed immediately. Tissue genomic DNAs (gDNAs) were extracted from tumor tissue using the QIAmp FFPE tissue kit (Qiagen, 56404, Germany). Both the cfDNA and gDNA concentrations were then quantified using Qubit dsDNA high-sensitivity (HS) assay kit (Invitrogen, Q32854, USA).
Library preparation, target region capture, and sequencing
The gDNAs extracted from tumor tissue samples were fragmented into 150 to 250 base pair (bp) pieces using a Covaris S220 (Covaris, S220, USA). The cfDNA and gDNA libraries were prepared using the Human EGFR, KRAS, BRAF, NRAS, PIK3CA gene mutation joint detection kit (Combinatorial Probe Anchor Synthesis; Beijing USCI Medical Devices Co., Ltd., JK001, China) according to the manufacturer’s instructions. A minimum of 20 ng of cfDNA was required as input. One to five libraries from the same sample type were pooled at an equal molar concentration and hybridized to a customized IDT Panel (~0.1 M). This panel was designed to enrich for selected coding and intronic regions of 20 cancer-related genes, including ALK, BRAF, DDR2, EGFR, ERBB2, FGFR1, JAK2, KRAS, MAP2K1, MET, NRAS, NTRK1, NTRK2, NTRK3, PIK3CA, POLE, RET, ROS1, STK11, and TP53 (see Supplementary Table 1). After PCR amplification, the quality and quantity of captured library were assessed using an Agilent 2100 bioanalyzer and ABI 7500 real-time PCR system (Life Technologies, 4351107, USA). The sequencing libraries were then loaded onto a high-throughput sequencing platform (USCISEQ-200, Beijing USCI Medical Devices Co., Ltd., China) to generate 100 bp pair-end reads.
Digital droplet PCR sequencing
The insertion and deletion (InDel) and single-nucleotide variant (SNV) assays specific for ddPCR (Bio-rad) were used to detect BRAF, EGFR, NRAS, KRAS, and PIK3CA mutations. The reaction mixture consisted of 10 μL ddPCR Supermix for Probes (Bio-Rad), 1 μL of Primers & Probe (FAM labeled + HEX labeled + Primers), DNA template (20 ng of input), and nuclease-free water to a total volume of 20 μL. The 20 μL of reaction mixture was then mixed with 70 μL of Droplet Generation Oil for Probes (Bio-Rad) using QD200 Droplet Generator (Bio-Rad) to generate an emulsion of droplets. The droplets were transferred using a pipette to a 96-well PCR plate, sealed with PCR plate seal foil, and amplified using C1000 Thermal Cycler (Bio-Rad) to the endpoint, using the following program: 95°C for 10 min, 40 cycles of 94°C for 30 s and 60°C for 1 min, one hold at 98°C for 10 min, and final holding at 4°C. Ramp rates were set to 2°C/s. After amplification, the plate was read on the QX200 Droplet Reader (Bio-Rad), and data analysis was performed using QuantaSoft Software version 1.7.4.0917 (Bio-Rad).
To determine the limit of detection (LOD), serial dilutions of mutant gDNA from 18 cell lines (WI38, SW480, NCI-H1975, LS-180, SW1271, A2058, NCI-H1650, NCI-H1355, NCI-H2087, HCT-15, SW48, NCI-H1573, A549, HCC2935, T84, NCI-H2122, GP5D, HCC4006) were prepared. The mutations in these cell lines were summarized in Supplementary Table 2. The dilutions were mixed with wild-type human gDNA to achieve a final gDNA concentration of 20 ng/μL and mutant frequencies of 1%, 0.3%, and 0.1%, with each dilution tested in eight replicates. The LOD was determined for each mutation class as the lowest mutant frequency that yielded a detection rate of at least 95% for the targeted mutations. The LOD was established at 0.1%.
Bioinformatics analysis
The raw reads with adapters were trimmed off. Leading and trailing low-quality (below 3) bases were removed and bases that average-quality below 15 within a sliding window of four bases were cut. Finally, reads with a length of above 50 bases were kept. The procedures above were performed using Trimmomatic (version 0.39). All clean sequence reads were mapped to human genomic reference sequences (hg19) using the Burrow-Wheeler Aligner MEM algorithm (version 0.7.12). Duplicated reads were marked using the Picard MarkDuplicates function (version 1.124). Local realignment of reads before variant calling was using GATK realignment function (version 3.4.46). Then, mutations were called by a single-sample mode of VarScan (version 2.4.1). Functional annotations were performed using ANNOVAR (version 20180416). Candidate mutations were filtered if (1) the depth was below 1000× in ctDNA and 800× in tissue, (2) reads with strand bias, (3) variant allele frequency (VAF) < 1.0% in tissue and < 0.2% in ctDNA.
Statistical analysis
Categorical variables were represented as counts (percentages). Continuous variables were summarized using the median and range. Pack-years were calculated by multiplying the number of packs of cigarettes smoked per day by the number of years. All the statistical analyses and graphics were performed using R (version 4.0.2).
Results
Clinicopathologic characteristics
This study reviewed the clinical characteristics of 173 lung cancer patients, and the results were summarized in Table 1. The median age of the cohort was 61 years (range, 25-82 years). Among the total patient population, 54.91% (95 out of 173) were male, while 45.09% (78 out of 173) were female. Stages I, II, III, and IV accounted for 6.36% (11 out of 173), 15.61% (27 out of 173), 15.61% (27 out of 173), and 62.43% (108 out of 173), respectively. A total of 83 patients (47.98%, 83 out of 173) were non-smokers, whereas 86 patients (49.71%, 86 out of 173) had a history of smoking. Among smokers, the median pack-year was 40 (range, 2.5-100). The results further revealed that the most common pathology was NSCLC, which constituted 98.27% (170 of 173) of the cases. Specifically, adenocarcinoma, squamous cell carcinoma, and adenosquamous carcinoma accounted for 78.03% (135 of 173), 15.03% (26 of 173), and 1.73% (3 of 173), respectively. 54.91% (95 of 173) of patients received at least one class of systemic therapy, and 45.09% (78 of 173) did not receive systemic therapy.
Clinical characteristics of study patients.
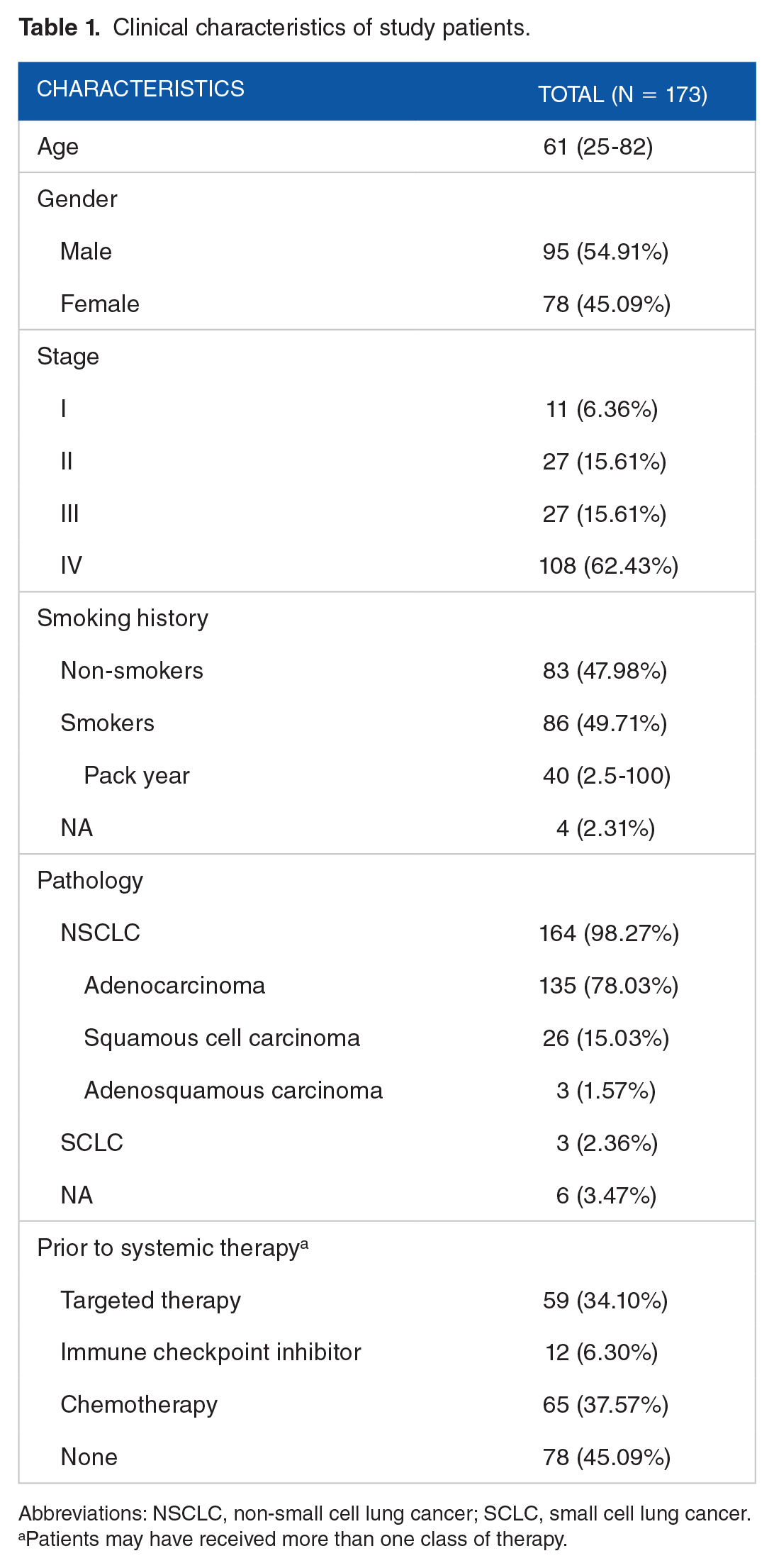
Abbreviations: NSCLC, non-small cell lung cancer; SCLC, small cell lung cancer.
Patients may have received more than one class of therapy.
Molecular profiling using ctDNA NGS assay
The median turnaround time for ctDNA NGS assay was 10 days (range 5-18 days). Out of the 173 ctDNA samples analyzed, we identified 247 ctDNA alterations in 61.85% (107 of 173) of the samples, resulting in an average of 1.43 alterations per sample. The ctDNA detection rates were 18.18% (2 of 11), 29.63% (8 of 27), 66.67% (18 of 27), and 73.15% (79 of 108) for Stages I, II, III, and IV patients, respectively. Regarding histologic types, the ctDNA detection rates were 51.55% (83 of 161) for adenocarcinoma and adenosquamous carcinoma, 57.69% (15 of 26) for squamous cell carcinoma, 100.00% (3 of 3) for SCLC, and 100% (6 of 6) for patients with unknown histology. The detected alterations included 104 SNVs, 134 insertions and deletions (InDels), four deletion-insertions (delins), three splicing mutations, and three gene rearrangements. EGFR (81 of 248, 32.66%) was the most frequently mutated gene, followed by TP53 (66 of 248, 26.61%). These mutated genes occurred in 35.83% (62 of 173) and 32.37% (56 of 173) of ctDNA samples, respectively. The EGFR exon 19 deletions, exon 20 insertion, L858R, and T790M were detected in 21, 1, 23, and 10 patients, respectively. The remaining mutated genes displayed frequencies below 15% in the ctDNA samples. Four SNVs and four deletions in the MET gene were detected, but none of these mutations located within MET exon 14. In addition, two BRAF mutations were detected, and they were all V600Es. We identified gene rearrangements in clinically relevant oncogene RET in two patients. The overview of alteration landscape is shown in Figure 2. The prevalence of NCCN guideline-recommended molecular alterations in NSCLC patients of our cohort is summarized in Table 2. Any NCCN-recommended molecular alteration (including all KRAS mutations, not limited to KRAS G12C) was detected in 40.85% of the NSCLC patient samples. Among four KRAS-mutated patients, one patient carried KRAS G12C mutation.

Alteration landscape of ctDNA from 173 lung cancer patients. The X-axis represents each sample and the Y-axis represents each mutated gene. Different colors represent different type of alteration and clinical characteristics. The top barplot shows the number of different alterations for each gene. ctDNA indicates circulating tumor DNA; Delins, deletion-insertion; InDel, insertion and deletion; SNV, single-nucleotide variant.
NSCLC patients with NCCN-recommended alterations based on ctDNA NGS results.

Abbreviations: NCCN, National Comprehensive Cancer Network; NSCLC, non-small cell lung cancer.
All KRAS mutations were included.
Concordance of detected alterations between tumor tissue and ctDNA
The concordance analysis was conducted in 107 lung cancer patients with matched tissue and ctDNA samples. Among these 107 tumor tissue samples, 210 alterations were identified in 95.33% (102 of 107) of the samples, resulting in an average of 2.07 alterations per sample. A total of 58 (54.21%, 58 of 107) patients carried EGFR mutations. Specifically, we identified 26 EGFR exon 19 deletions, two exon 20 insertions, 27 L858Rs, and nine T790Ms. In addition, four SNVs and one deletion were detected in the MET gene, and one of these mutations was located within MET exon 14. Furthermore, of the five BRAF mutations detected, two were V600Es. The tumor tissue NGS assay also identified two ALK rearrangements and two RET rearrangements. The overview of alteration landscape of 107 tumor tissue samples is shown in Figure 3A.
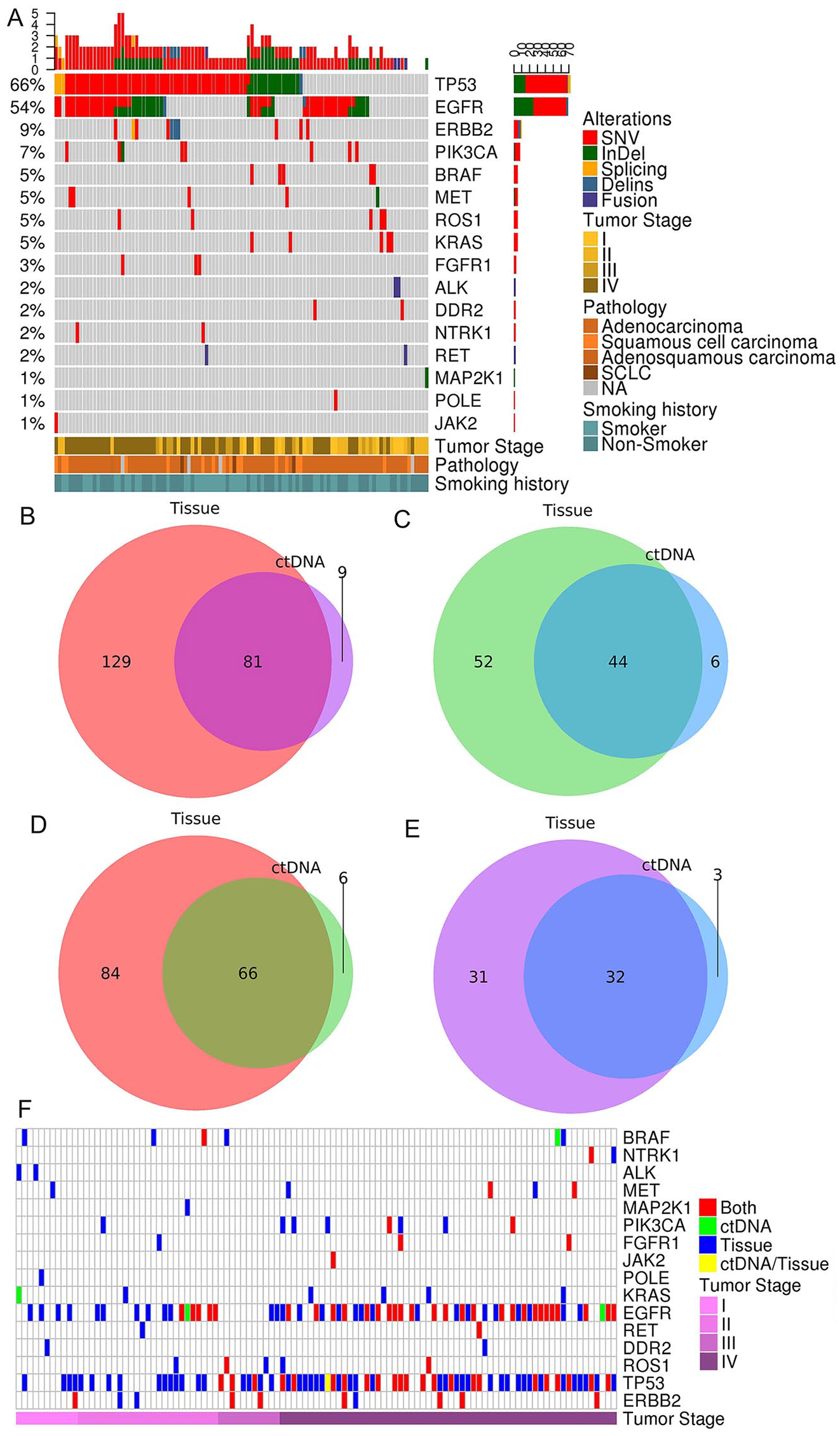
Molecular profiles detected in 107 paired tumor tissue and ctDNA samples. (A) alteration landscape of tumor tissue from 107 lung cancer patients. The X-axis represents each sample and the Y-axis represents each mutated gene. Different colors represent different type of mutation and clinical characteristics. The top barplot shows the number of different alterations for each gene. Concordance of (B, D) all alterations and (C, E) NCCN-recommended alterations in (B, C) all paired tissue and ctDNA samples and advanced NSCLC paired tissue and ctDNA samples. Venn diagrams representing the numbers of alteration counts detected in tissue (left circle), ctDNA (right circle) for (B, D) all alterations and (C, E) NCCN-recommended alterations. (F) Heatmap of alteration profiles across 15 detected genes with at least one detected alteration in tissue or ctDNA. Red bar: concordant gene alteration in two tests; green bar: gene alteration in ctDNA only; blue bar: gene alteration in tissue only; yellow bar: gene alteration both in ctDNA and tissue, but alteration at amino acid substitution level differs. ctDNA indicates circulating tumor DNA; Indels, insertions and deletions; SNVs, single-nucleotide variants.
The median time interval between tissue and blood draw was 4 days, ranging from 0 to 7 days. Across these 107 paired tumor tissues and ctDNAs, a total of 300 alterations were detected by tissue and ctDNA NGS tests. Of these 300 alterations, 81 were shared between the two sample types (see Figure 3B), including 18 InDels, 62 SNVs, and one gene rearrangement. The concordance rate was 34.62% (18 of 52) for InDels, 38.04% (62 of 163) for SNVs, and 25.00% (1 of 4) for gene rearrangements. About 38.57% (81 of 210) of tissue alterations could be detected in paired ctDNAs, and 90.00% (81 of 90) of ctDNA alterations could be detected in paired tissues. The overall concordance rate was 36.99% (81 of 219). The sensitivity of ctDNA NGS assay was 38.57% for alterations at the amino acid substitution level. At patient level, among 99 alteration-positive patients, 50.51% (50 of 99) patients had at least one concordant alteration in ctDNA. The concordance rate was 14.43% and 43.77% at amino acid substitution level and 21.21% and 62.32% at patient level for early-stage and advanced lung cancer patients, respectively. As for NCCN-recommended alterations, 96 and 50 NCCN-recommended alterations were identified by tumor tissue and ctDNA NGS assay, respectively. A total of 44 alterations were shared by two assays, and the concordance rate was 42.72% (44 of 103; see Figure 3C). The concordance rate was 31.11% and 48.61% at amino acid substitution level and 33.33% and 56.00% at patient level for early-stage and advanced lung cancer patients, respectively. The detected results of tumor tissue and ctDNA NGS assays were summarized in Supplementary Table 3. Among 81 concordant alterations, VAFs of 81.48% (66 of 81) mutations in tumor tissues were higher than ctDNAs (see Supplementary Figure 1).
There were 64 advanced (Stage III and IV) NSCLC patients. A total of 222 alterations were identified in these 64 paired tumor tissues and ctDNAs. When examining the concordance between tissue and ctDNA NGS assay in the subset of patients with advanced NSCLC, we observed an overall agreement rate of 42.31% (66 of 156; see Figure 3D). There were 66 alterations were shared by two assays, including 14 InDels, 51 SNVs, and one gene rearrangement. About 44.00% (66 of 150) of tissue alterations could be detected in paired ctDNAs, and 91.67% (66 of 72) of ctDNA alterations could be detected in paired tissues. Among the 63 tumor tissue samples that had at least one genetic alteration identified, 58.73% (37 of 63) of the corresponding ctDNA samples also had at least one concordant alteration detected. As for NCCN-recommended alterations, 63 and 35 alterations were identified in 48 tumor tissues and 26 ctDNAs, respectively. Thirty-two alterations were shared by two assays, and the concordance rate was 48.48% (32 of 66; see Figure 3E). In 54.17% (26 of 48) of tissue-positive advanced NSCLC patients, the paired ctDNA samples contained at least one concordant NCCN-recommended alteration. The concordance rate was 31.11% and 33.33% at amino acid substitution level and patient level for early-stage NSCLC patients, respectively. Four advanced NSCLC patients carried NCCN-recommended alterations in ctDNA only. In addition to tissue biopsy, ctDNA NGS assay increased the detection rate by 12.00% (3 of 25).
Alterations within 16 genes were detected by the NGS assay (see Figure 3F). A gene was considered concordant if at least one concordant alteration was detected within this gene. At the gene level, for the 20 lung cancer-related genes in targeted regions, the concordance rate was 55.00% (11 of 20). EGFR was the most concordant gene in our analysis, with 38 concordant EGFR mutations spread in 28.04% of samples, followed by TP53, with 24 concordant TP53 mutations spread in 21.50% of samples. 53.27% (57 of 107) of patients had no concordance among these 20 genes.
Concordance of detected or undetected mutations between ddPCR and NGS
The ddPCR provides precise, highly sensitive quantification of nucleic acids. We used ddPCR to examine a subset of mutations that were detected or undetected by the ctDNA NGS assay. Of the 173 patients, 82 had sufficient DNA remaining after ctDNA NGS testing to permit ddPCR testing. A total of 176 mutations that were either detected or not detected in ctDNA were selected and verified using ddPCR. The results of 175 mutations were consistent between ddPCR and ctDNA NGS assay. A total of 30 mutations detected by ctDNA NGS test were also identified by ddPCR, and 145 undetected by ctDNA NGS test were also negative in the ddPCR analysis (see Figure 4A). Among these concordant mutations, 132 were SNVs, while 43 were deletions. The concordance rates for SNVs and deletions were 100% (132 of 132) and 97.73% (43 of 44), respectively. At the gene level, KRAS, PIK3CA, BRAF, and NRAS exhibited a high consistency of 100% between ddPCR and ctDNA tests (see Figure 4B). However, one EGFR exon 19 deletion with a VAF of 0.12% was undetected by ctDNA, as it did not surpass the detection limit of the ctDNA test. This deletion was subsequently verified by ddPCR. Overall, the results demonstrated a high level of agreement between ddPCR and ctDNA tests. The ctDNA NGS exhibited a sensitivity of 96.67% and a specificity of 100%.
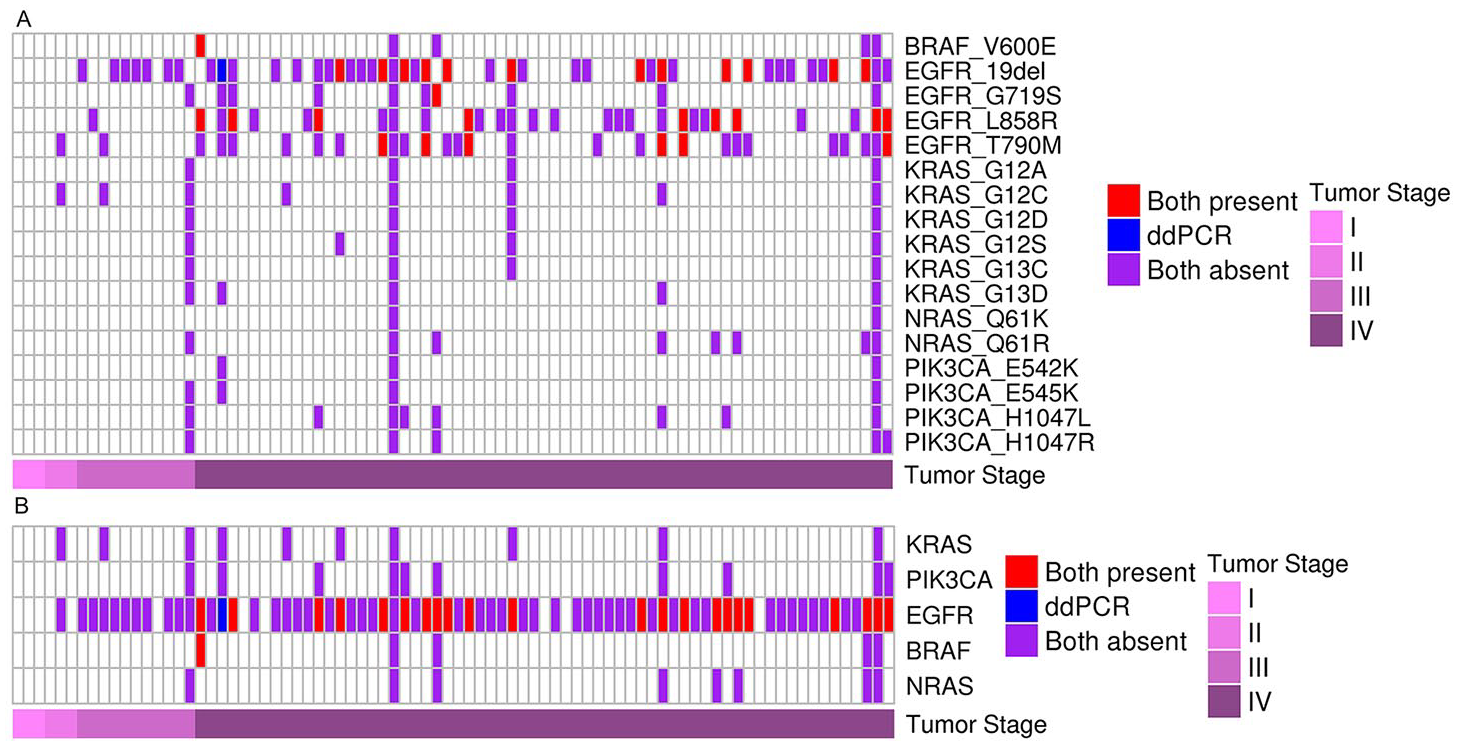
Comparison of mutation profiles characterized by ddPCR and ctDNA at the (A) amino acid substitution level and the (B) gene level. Only 82 patients had sufficient remaining DNA to enable the subsequent ddPCR and are represented in the figure. Red: mutation detected by ddPCR and ctDNA; blue: mutation only detected by ddPCR; purple: mutations undetected both by ddCPR and ctDNA. ctDNA indicates circulating tumor DNA; ddPCR, droplet digital PCR.
ctDNA and tumor heterogeneity
Patient 01015, a 66-year-old male diagnosed with stage IV adenocarcinoma and a 40-year history of smoking half a pack per day, exhibited concomitant EGFR T790M/C797S mutations in cis, which were detected in both tumor tissue and ctDNA. Notably, the ctDNA assay identified the EGFR C797S c.T2389A mutation, while the tissue biopsy identified the EGFR C797S c.G2390C mutation (see Table 3).
Detected mutations in tissue and ctDNA from two patients.
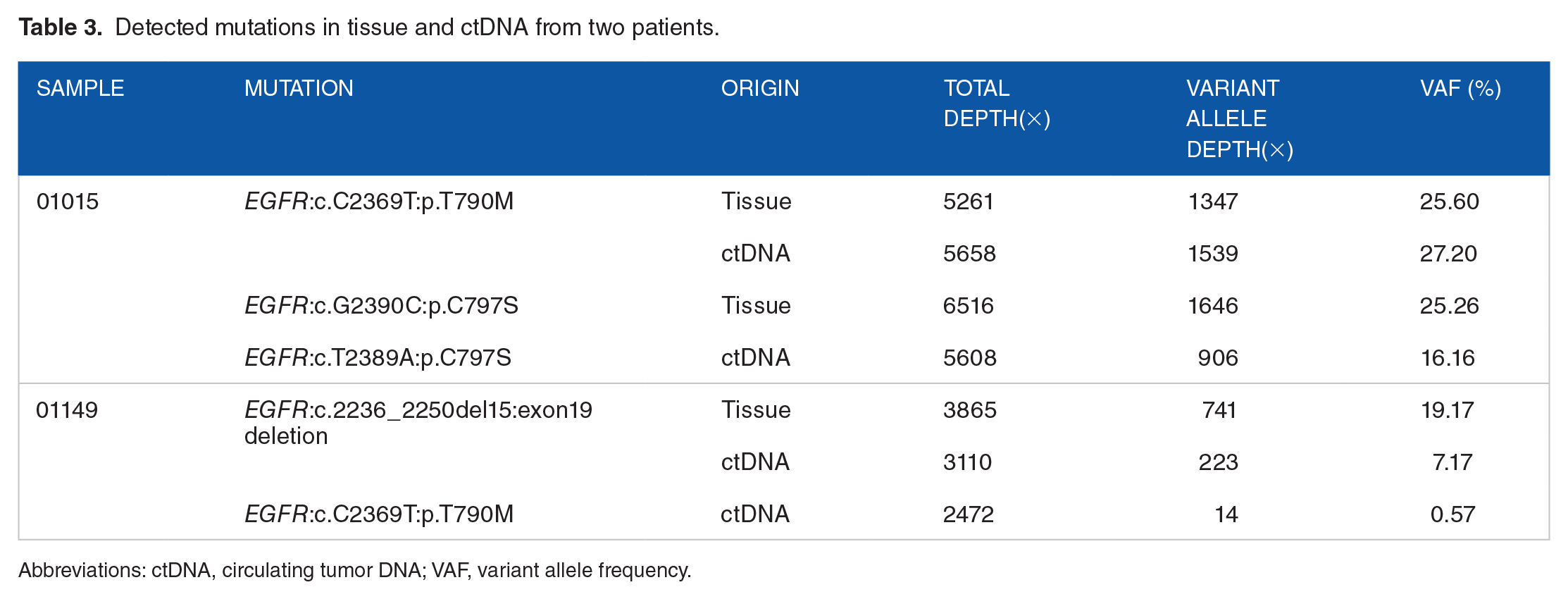
Abbreviations: ctDNA, circulating tumor DNA; VAF, variant allele frequency.
Patient 01149, a 51-year-old male diagnosed with stage IV squamous cell carcinoma and no smoking history, harbored an EGFR exon 19 deletion mutation, as identified by the tumor tissue biopsy. This patient received gefitinib, a first-generation EGFR tyrosine kinase inhibitor (TKI), as part of the therapeutic regimen. Subsequently, the patient developed drug resistance, and no drug-resistant variants were identified by tissue biopsy. However, ctDNA assay detected the presence of EGFR T790M with a VAF of 5.7% (see Table 3). The results of tumor tissue and ctDNA NGS assays of these two patients were confirmed by ddPCR (see Supplementary Figures 2–6).
The treatment response to targeted therapy based on ctDNA NGS results
We reviewed the treatment response of 17 lung cancer patients who received EGFR-TKIs therapy. The ctDNA NGS genotyping identified EGFR actionable mutations in 17 samples and led these patients to EGFR-TKIs therapy. The median age of these 17 patients was 62 years (range, 42-74 years). Most of the patients were at stage IV (9 of 12, 75%). Partial response was achieved in 12 patients, and stable disease was achieved in 5 patients. The EGFR exon 19 deletion was identified in eight patients, and two of them were co-existed with EGFR exon 20 T790M. Six patients with EGFR exon 19 deletion mutation were treated with icotinib and achieved partial response. Two patients with concurrent EGFR exon 19 deletion and EGFR exon 20 T790M mutations received osimertinib and achieved partial response and stable disease, respectively. EGFR exon 21 L858R was identified in nine patients, and two of them were co-existed with EGFR exon 20 T790M. Seven patients with EGFR exon 21 L858R were matched to icotinib, and five patients achieved partial response and two patients achieved stable disease. Two patients with concurrent EGFR exon 21 L858R and EGFR exon 20 T790M were matched to osimertinib and achieved stable disease and partial response, respectively (see Table 4).
Patients matched to targeted therapy base on ctDNA NGS results and treatment response.

Abbreviations: ctDNA, circulating tumor DNA; PR, partial response; SD, stable disease.
Discussion
Targeted therapies for lung cancer patients are typically administered based on the molecular profile of the tumor. However, tumor tissue biopsy poses the risk of trauma and associated complications for patients, and acquiring sufficient tumor tissue for molecular profiling presents challenges. In addition, the genetic testing of a single tissue biopsy only provides a limited snapshot of the tumor molecular profile due to tumor heterogeneity. In this context, the emergence of ctDNA as a promising noninvasive approach for profiling lung cancer tumors has garnered considerable attention.
In our study, we made several observations regarding the mutation landscape revealed by ctDNA compared with tumor tissue. We found that the mutation profiles of EGFR were highly concordant between ctDNA and tissue biopsy, indicating the reliability of ctDNA in capturing key mutations. Notably, a significant proportion (89.01%) of ctDNA-derived mutations were also identified in paired tissue samples, highlighting the potential of ctDNA as a viable alternative to invasive tissue biopsy. However, there were discrepancies in mutation detection between ctDNA and tissue samples, with only 38.57% of tumor-derived mutations being detected in ctDNA, which suggests ctDNA NGS assay exhibits certain limitations in its ability to comprehensively capture the full spectrum of genomic alterations within the tumor. This is due to the low concentrations of mutant DNA fragments in ctDNA. We found that for the concordant alterations detected in both tumor tissue and ctDNA samples, most (81.48%) had a lower VAF in the plasma compared with the corresponding tumor tissue. This suggests that the tumor-derived mutations become diluted when entering the peripheral circulation, resulting in some alterations being undetectable in the plasma. In addition, our samples were collected following the patients’ potential receipt of systemic therapies, which may also have influenced the ctDNA content in the blood. This is an important consideration, as prior treatments could lead to a decrease in the amount of tumor-derived DNA circulating in the peripheral blood. This scenario also aligns with the practical clinical reality, where patients may undergo ctDNA assays following treatment administration to evaluate the efficacy of the therapy. The time interval between tissue and blood draw has been extensively investigated as a factor impacting the concordance between tissue and liquid biopsy. It has been observed that as the time intervals increase, the concordance decreases.15-17 This decrease in concordance with longer time intervals aligns with the concept of tumor evolution over time. We restricted the time intervals between tissue biopsy and liquid biopsy to within 7 days to minimize the potential confounding effect of temporal factors on the observed concordance.
The results demonstrated that the detection rate of ctDNA and the concordance between tumor tissue and ctDNA NGS assay were both significantly higher in patients with advanced lung cancer compared to those with early-stage lung cancers. In comparison to patients with advanced lung cancer, the use of ctDNA assays in early-stage disease settings faces greater challenges in comprehensively capturing tumor heterogeneity, thereby limiting its direct utility in guiding treatment decisions for these patients. In contrast to the late-stage setting, treatment options for early-stage patients are relatively limited, primarily focused on surgical resection. At this earlier phase of disease, the direct utility of ctDNA analysis in guiding adjustments to the therapeutic approach may be more constrained. However, ctDNA testing results could potentially assist in the prognostic evaluation and monitoring of disease recurrence in early-stage patients. Timely identification of potential recurrence risks could prompt more proactive surveillance strategies and facilitate the judicious administration of adjuvant therapies. If ctDNA detection can reliably identify specific driver gene mutations in the early-stage setting, it may provide a basis for informing individualized treatment decisions in the future. Looking forward, if the sensitivity and specificity of ctDNA detection can be enhanced in the early-stage setting, and if it can be integrated with other relevant biomarkers, the technology may have the potential to inform individualized treatment decision-making for patients with early-stage lung cancer. However, further clinical validation studies will be required to fully establish the feasibility and utility of this approach.
We validated a group of ctDNA-detected and -undetected mutations using ddPCR. There was an extremely high concordance of 99.43% (175 of 176) between ddPCR and ctDNA NGS assay. Similar results were also reported in other studies. Stitz et al 18 reported a concordant rate of 91% between ctDNA NGS and ddPCR in 28 lung cancer plasma samples. In another study, 13 driver mutations in 13 selected patients detected in ctDNA were all validated using ddPCR. 19 Yang et al 20 evaluated the concordance between the results of ctDNA NGS assay and ddPCR in 42 plasma samples and the coincidence rate for positive and negative mutations was 97.44% and 97.30%, respectively. These results have important clinical significance, demonstrating that ctDNA can accurately detect the mutations in the peripheral blood. Compared to one mutation that ddPCR detected in one single test, NGS-based ctDNA assay allows simultaneous analysis of a wide range of mutations of interest to researchers.
The ctDNA assay detected the EGFR C797S c.T2389A mutation, while the tissue biopsy detected the EGFR C797S c.G2390C mutation in patient 01015. The co-occurrence of the EGFR T790M/C797S mutations in cis is particularly notable. The EGFR C797S is known to confer resistance to third-generation EGFR-TKIs, such as osimertinib, which are designed to overcome the T790M-mediated resistance.21,22 The presence of both mutations indicated a potential dual resistance mechanism, making the tumor even more challenging to treat effectively with existing targeted therapies. Moreover, the presence of multiple mutations within the tumor indicates that different subclones of cancer cells may have developed different resistance mechanisms. Understanding tumor heterogeneity is crucial for guiding treatment decisions and selecting appropriate therapeutic strategies to address the diverse genetic alterations present within the tumor. An illustrative example of clinical relevance of ctDNA assay was observed in patient 01149, who initially responded to gefitinib targeting the EGFR exon 19 deletion but developed therapeutic resistance after a year of treatment. The ctDNA NGS assay identified the secondary resistance mechanism of EGFR exon 20 T790M mutation, which was missed in the tissue biopsy. This case demonstrated that ctDNA assays could partially overcome tumor heterogeneity and provide valuable insights into therapeutic resistance mechanisms that may be missed by tissue biopsy alone.
Among 17 patients with positive EGFR mutations in ctDNAs who received matched EGFR-TKIs therapies tailored based on ctDNA NGS results, 12 patients achieved a partial response and two patients had stable disease. These results highlight the potential of ctDNA assay in guiding patients toward personalized targeted therapy, particularly for EGFR-addicted tumors. Despite the benefits of TKIs in targeted therapy for lung cancer patients, acquired resistance to targeted therapy typically arises within 1 to 2 years of treatment initiation, necessitating multiple biopsies to identify genomic alterations and guide patients toward appropriate targeted therapies. In four patients with acquired TKI resistance EGFR T790M mutation detected in ctDNA, treatment with osimertinib resulted in partial response for two patients and stable disease for the other two. Furthermore, the ctDNA NGS assay results of patient 01149 demonstrated the assay’s ability to detect acquired resistance mutations by overcoming the challenges posed by tumor heterogeneity. In addition, due to the ability for multiple collections through blood draw, ctDNA has the potential for dynamic monitoring of treatment response. Regular analysis of ctDNA facilitates the timely detection of mutations and changes in resistance mechanisms that may arise during treatment, thereby providing valuable insights for guiding adjustments to treatment plans.
The ctDNA NGS assay has certain limitations. First, regarding sensitivity, the current NGS platform boasts over 99% sensitivity at VAFs of 1% and above,23,24 while our approach exhibits a remarkable sensitivity of over 95% even at VAFs as low as 0.2%. Despite this, there are still many tissue-derived variations that remain undetected. Second, the specificity of this technology is also limited, and there may be false positive results, especially when detecting low-abundance variants. 25 Therefore, prior to employing ctDNA NGS detection, it is crucial to validate the performance of the detection assay to prevent potential false positives or false negatives from misleading clinical decisions. Carefully considering these limitations will help ensure appropriate clinical interpretation and guide the responsible application of this technology in patient care.
There are also certain limitations to this study. The sample size in this study was relatively small, with only 107 patients used to compare the concordance between tumor tissue and ctDNA NGS analysis. A larger, more diverse patient cohort would be necessary to fully characterize the performance of this ctDNA approach across different disease stages and pathologies. In addition, most of our study subjects had NSCLC, with very few small cell lung cancer patients. In the future, it will be important to expand the small cell lung cancer cohort to conduct more extensive analysis. The study population also included both treatment-naïve and previously treated patients. The treatment status of the patients may significantly impact ctDNA levels in the peripheral blood. Therefore, it will be crucial to perform subgroup analyses for the treatment-naïve and previously treated patient subsets. In future studies, expanding the overall study population and conducting these subgroups comparisons would be valuable. While this study did include early-stage lung cancer samples and explored the application of ctDNA NGS in early disease, a more comprehensive analysis focused on a large cohort of advanced lung cancer patients is also warranted.
Conclusions
In conclusion, our study supports the potential of ctDNA analysis as a noninvasive supplementary to tissue biopsies for mutation profiling in advanced lung cancer patients. The case studies presented in our research highlight the clinical significance of specific mutations detected in ctDNA and emphasize the need for comprehensive mutation profiling and monitoring of resistance mechanisms to guide treatment decisions and improve patient outcomes. Moreover, our study demonstrates the clinical impact of ctDNA NGS genotyping in guiding the administration of targeted therapies. Complementing tissue biopsy with ctDNA assay could increase access to targeted therapies. These findings contribute to the growing body of evidence supporting the value of ctDNA analysis in advanced lung cancer management.
Supplemental Material
sj-docx-1-onc-10.1177_11795549241285238 – Supplemental material for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers
Supplemental material, sj-docx-1-onc-10.1177_11795549241285238 for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers by Kun Li, Nana Zhang, Bing Xu, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Guangyu Shan, Sihang Gao, Bo Yu, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang, Nanying Che and Xiaoyong Ji in Clinical Medicine Insights: Oncology
Supplemental Material
sj-docx-2-onc-10.1177_11795549241285238 – Supplemental material for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers
Supplemental material, sj-docx-2-onc-10.1177_11795549241285238 for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers by Kun Li, Nana Zhang, Bing Xu, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Guangyu Shan, Sihang Gao, Bo Yu, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang, Nanying Che and Xiaoyong Ji in Clinical Medicine Insights: Oncology
Supplemental Material
sj-docx-3-onc-10.1177_11795549241285238 – Supplemental material for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers
Supplemental material, sj-docx-3-onc-10.1177_11795549241285238 for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers by Kun Li, Nana Zhang, Bing Xu, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Guangyu Shan, Sihang Gao, Bo Yu, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang, Nanying Che and Xiaoyong Ji in Clinical Medicine Insights: Oncology
Supplemental Material
sj-docx-4-onc-10.1177_11795549241285238 – Supplemental material for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers
Supplemental material, sj-docx-4-onc-10.1177_11795549241285238 for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers by Kun Li, Nana Zhang, Bing Xu, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Guangyu Shan, Sihang Gao, Bo Yu, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang, Nanying Che and Xiaoyong Ji in Clinical Medicine Insights: Oncology
Supplemental Material
sj-docx-5-onc-10.1177_11795549241285238 – Supplemental material for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers
Supplemental material, sj-docx-5-onc-10.1177_11795549241285238 for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers by Kun Li, Nana Zhang, Bing Xu, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Guangyu Shan, Sihang Gao, Bo Yu, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang, Nanying Che and Xiaoyong Ji in Clinical Medicine Insights: Oncology
Supplemental Material
sj-docx-6-onc-10.1177_11795549241285238 – Supplemental material for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers
Supplemental material, sj-docx-6-onc-10.1177_11795549241285238 for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers by Kun Li, Nana Zhang, Bing Xu, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Guangyu Shan, Sihang Gao, Bo Yu, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang, Nanying Che and Xiaoyong Ji in Clinical Medicine Insights: Oncology
Supplemental Material
sj-xlsx-7-onc-10.1177_11795549241285238 – Supplemental material for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers
Supplemental material, sj-xlsx-7-onc-10.1177_11795549241285238 for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers by Kun Li, Nana Zhang, Bing Xu, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Guangyu Shan, Sihang Gao, Bo Yu, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang, Nanying Che and Xiaoyong Ji in Clinical Medicine Insights: Oncology
Supplemental Material
sj-xlsx-8-onc-10.1177_11795549241285238 – Supplemental material for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers
Supplemental material, sj-xlsx-8-onc-10.1177_11795549241285238 for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers by Kun Li, Nana Zhang, Bing Xu, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Guangyu Shan, Sihang Gao, Bo Yu, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang, Nanying Che and Xiaoyong Ji in Clinical Medicine Insights: Oncology
Supplemental Material
sj-xlsx-9-onc-10.1177_11795549241285238 – Supplemental material for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers
Supplemental material, sj-xlsx-9-onc-10.1177_11795549241285238 for Utility of Circulating Tumor DNA Assay in Identifying Mutations and Guiding Matched Targeted Therapy in Lung Cancers by Kun Li, Nana Zhang, Bing Xu, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Guangyu Shan, Sihang Gao, Bo Yu, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang, Nanying Che and Xiaoyong Ji in Clinical Medicine Insights: Oncology
Footnotes
Acknowledgements
We thank all the patients who participated in this study, and colleagues who contributed to this work.
Author Contributions
Nanying Che and Xiaoyong Ji designed the study. Kun Li, Nana Zhang, Zichen Liu, Dan Zhao, Yujie Dong, Jing Mu, Haifeng Lin, Xiaoxi Pan, Yanrong Wang, Dongxing Zhang were responsible for sample collection and experiments. Bing Xu, Sihang Gao, Guangyu Shan, Bo Yu headed all computational and analytical aspects. Kun Li, Nana Zhang, Bing Xu wrote the article, and all authors participated in proofreading. All authors contributed to editorial changes in the article. All authors read and approved the final article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Beijing Municipal Science and Technology Project, grant number Z181100001918027, Z191100006619079.
Availability of Data and Materials
The datasets generated and analyzed during this study are available from the corresponding author on a reasonable request.
Ethics Approval and Consent to Participate
This study was conducted in accordance with the Declaration of Helsinki and was approved by the institutional review board of the Beijing Chest Hospital (approval number: 2020[25-01], approval date: September 16, 2020). Written informed consent was obtained from all subjects in the study.
Patient Consent for Publication
All patients have signed patients’ informed consent.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.