
Abstract
Gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs) are a heterogeneous group of neoplasms with an increasing incidence in the last few decades. Despite therapeutic advances in the management of GEP-NENs, resistance to many of these treatments has made their management a great challenge. One of the most recent advances in oncologic therapy is targeting multiple receptors simultaneously and engaging immune cells in the tumor microenvironment through bispecific antibodies (BsAbs). Since the FDA approval of the anti-CD3 × anti CD19 BsAb blinatumomab, for management of B-cell acute lymphoblastic leukemia, around a hundred different BsAbs have been developed and tested in various clinical trials. In this article, we review the current development of BsAbs developed or being currently tested for the management of GEP-NENs, their mechanism of action, current results from ongoing trials, toxicities, and upcoming trials.
Introduction
Gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs) are heterogeneous neoplasms originating from neuroendocrine secretory cells of the gastroenteropancreatic system. Their incidence has risen in the last decade from 1.9 cases per 100 000 to 6.8 cases per 100 000 in the United States. They are classified according to their histological differentiation into well-differentiated neuroendocrine tumors (NETs) that are divided into 3 grades based on their proliferative activity, and poorly differentiated neuroendocrine carcinomas (NECs) that are, by definition, high grade. 1 Despite the significant increase in incidence, there has only been a modest improvement in therapeutic outcome. This is especially the case for the high-grade well-differentiated and the poorly differentiated subgroups. Resistance to conventional therapies is attributed to various mechanisms including, but not limited to, epigenetic modification, protein expression alteration, somatostatin receptor (SSTR) internalization/phosphorylation, and mutations at drug binding sites. 2 One example of an emerging novel therapy for several cancers is bispecific antibodies (BsAbs). BsAbs are antibodies with 2 binding sites that target either 2 different antigens or 2 different epitopes on the same antigen, which leads to an increased therapeutic effect over monoclonal antibodies that have a single target. They have variable mechanisms of action, including connecting immune cells to the tumor cells. BsAbs additionally target different tumor antigens and are able to block dual signaling pathways leading to cytotoxicity. 3
First described by Nisonoff and co-workers more than 50 years ago, 4 BsAbs can be divided based on their function into BsAbs that act directly on their targets by neutralizing/activating them, and BsAbs that target specificity to deliver a therapeutically active moiety, such as a toxin. BsAbs tend to be either “IgG-like,” large molecules containing an Fc region and its related effector function while resembling conventional antibodies and having a longer serum half-life, or “non-IgG-like,” lacking the Fc region as, smaller molecules that exhibit enhanced tissue penetration but a shorter half-life. Over 100 BsAb formats have been developed into technology platforms and are in clinical use to treat multiple hematological and solid tumors.5,6
This review article will summarize the current advances and discuss future directions for the role of BsAbs in management of GEP-NENs (Table 1).
Current bispecific antibodies in management of GEP-NENs.
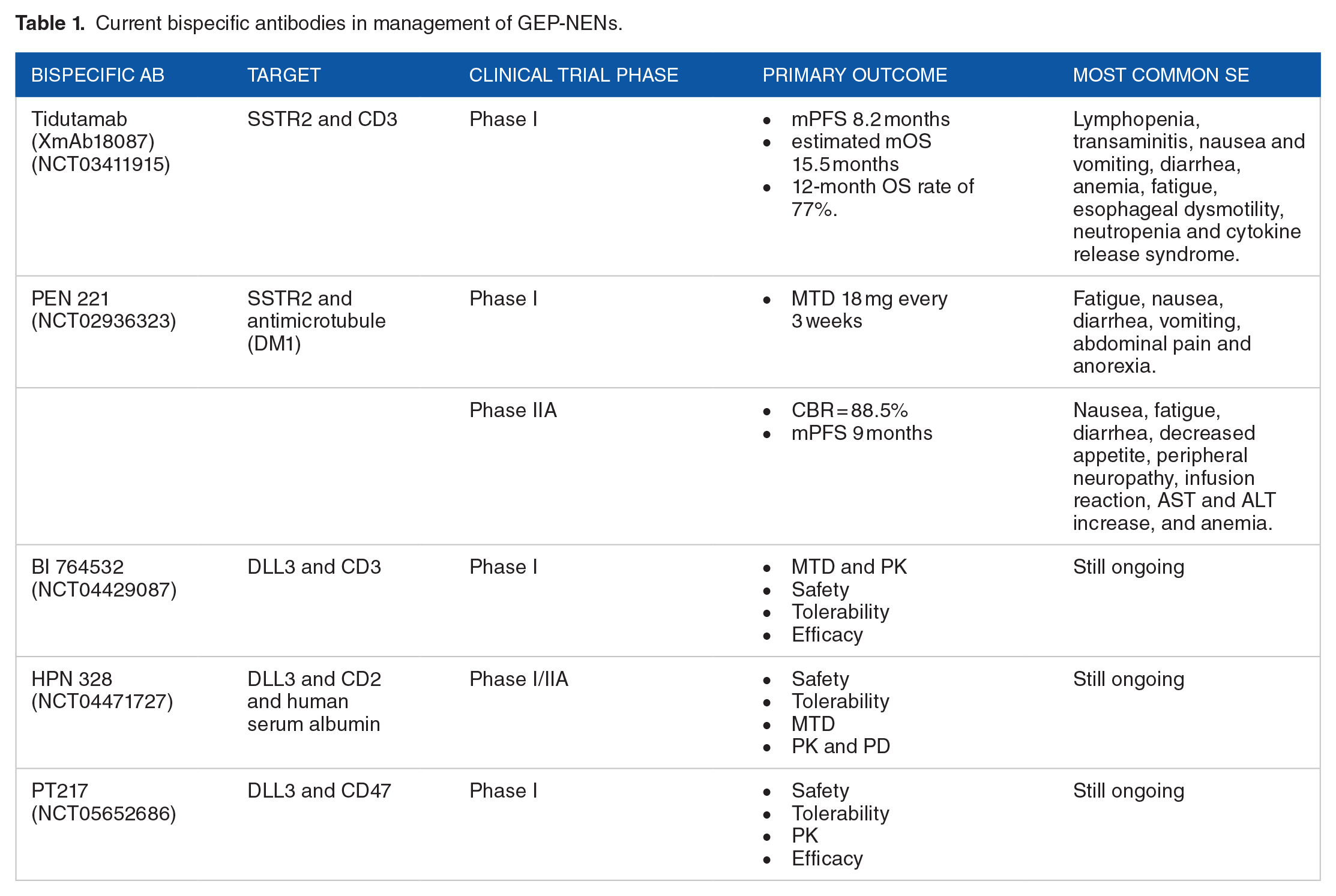
Current Bispecific Antibodies in GEP-NETS
Tidutamab (XmAb18087) (SSTR2 × CD3)
The majority of well-differentiated GEP-NETs express SSTRs predominantly SSTR2, which is the target of multiple therapies, specifically somatostatin analogues (SSAs) and peptide receptor radionuclide therapy (PRRT).7 -9 Tidutamab is an example of an anti-SSTR2 × anti-CD3 bispecific antibody that contains both an SSTR2 binding domain and a T-cell binding domain (CD3) (Figure 1). 10 It was strategically designed to apply the efficacy of T-cell immunotherapy in SSTR2-expressing malignancies. Preclinical studies demonstrated significant T-cell activation and killing of SSTR2-expressing cancer cells in vitro and induced anti-tumor activity in human PBMC-engrafted mice in vivo. 11 These results led to the development of a phase I clinical trial to evaluate the safety and tolerability of Tidutamab in patients with advanced, well-differentiated NETs. The study included patients with locally advanced or metastatic grade 1 or 2 NETs of different origins (pancreatic, GI, lung, or undetermined origin) that had progressed on at least 2 lines of therapies. Forty-two patients were enrolled in the trial; 41 of those received Tidutamab. The majority of patients were grade 2 (57.9%) and pancreas was the most common primary tumor (46.3%) followed by midgut tumors (22.0%) and lung NETs (19.5%). It is important to note that most of the patients had progressed on SSAs (41.5%) and PRRT (50%). Preliminary results reported that among 41 of the subjects, there was a median progression-free survival of 8.2 months (95% CI 2.7, 14.3) with estimated median overall survival (OS) of 15.5 months and 12-month OS rate of 77%. The best response was stable disease (SD; 26.8%) with no complete or partial responses observed. Interestingly, higher PD-L1 expression was associated with shorter survival time on the study. The most common reason for discontinuing treatment was disease progression (n = 17), followed by toxicity (n = 13), withdrawn consent (n = 7), physician decision (n = 1), loss to follow-up (n = 1), or another undisclosed reason (n = 1). Regarding safety profile, 65.9% of patients experienced at least 1 adverse effect (AE). The most frequent grade 3 or 4 treatment-related AE included lymphopenia (29.0%), transaminitis (19.5%), nausea and vomiting (17.1%), diarrhea (9.8%), anemia (7.3%), fatigue (7.3%), esophageal dysmotility (7.3%), neutropenia (4.9%), and cytokine release syndrome (CRS; 4.9%). No treatment-related deaths were observed. 12 Given the modest clinical benefit of Tidutamab with no radiological response and limited progression-free survival (PFS) compared with other targeted therapies, there is no further development currently for refractory GEP-NETS.

Mechanism of action for tidutamab. 10 (A) Tidutamab composed of an anti-SSTR2 on tumor cell and anti-CD3 found on T-cells. (B) When activated, SSTR and CD3 trigger T-cell-mediated cytotoxicity.
PEN 221 (SSTR2 × DM1)
Another promising targeted SSTR2-mediated drug for SSTR2-expressing tumors is PEN-221. As an antibody drug conjugate (ADC), PEN-221 combines an SSTR2 agonist molecule with a cytotoxic payload DM1 which is the microtubule targeting agent maytansinoid emtansine (Figure 2). 13 PEN-221 selectively binds with high affinity to SSTR2 resulting in the accumulation of the cytotoxic DM1, leading to cell cycle arrest and apoptosis. 14 Preclinical data showed complete regression in multiple SSTR2 xenograft mouse models. A phase I clinical trial using PEN-221 in 23 patients with NETs and SCLC, 13 demonstrated that PEN-221 was well tolerated with evidence of antitumor activity, and the maximum tolerated dose (MTD) was established at 18 mg every 3 weeks. 3 patients with gastrointestinal or pancreatic NETs had PR, while 11 patients had SD at 9 weeks, with 8 sustaining for 18 to 45 weeks. The most frequent adverse events were fatigue (48%), nausea (48%), diarrhea (44%), vomiting (26%), abdominal pain (26%), and anorexia (22%).A Phase IIA expansion cohort tested PEN-221 in patients with advanced SSTR2-positive midgut NETs, pNETs, and SCLC. Thirty-two patients (25 PRRT naïve patients; 7 PRRT-treated recurrent patients) were enrolled between January 2018 and June 2020. The regimen for MTD was changed from 18 mg (in the first 9 patients) to 8.8 mg/m2 in the remainder of the patients. Achieving more uniform exposures and reducing toxicity in patients with lower body-surface-area (BSA), the results recorded that PEN-221 was well tolerated in all patients given the dose of 8.8 mg/m2. Of the 26 patients that were evaluable for response, 23 (88.5%) had SD reported as their best response. This presented a clinical benefit rate (CBR) of 88.5% (CI 69.8%-97.6%) with no patients having CR or PR. As for target lesions, shrinkage was observed in 10 (38%) patients. Median progression-free survival (mPFS) for this cohort was 9 months (CI 5-16.5 months). As for the most frequent treatment-related adverse events, they were recorded as nausea (50%), fatigue (47%), diarrhea (47%), decreased appetite (47%), peripheral neuropathy (34%), infusion reaction (31%), aspartate transaminase (AST) and alanine transaminase (ALT) increase (28% and 22%), and anemia (25%). Only 11 (34%) of these events were ⩾ grade 3. The study overall concluded that PEN-221 was well tolerated and demonstrated efficacy at 8.8 mg/m2q 3 weeks and therefore a randomized trial of PEN-221 in GI midgut NET patients is currently in development. 15
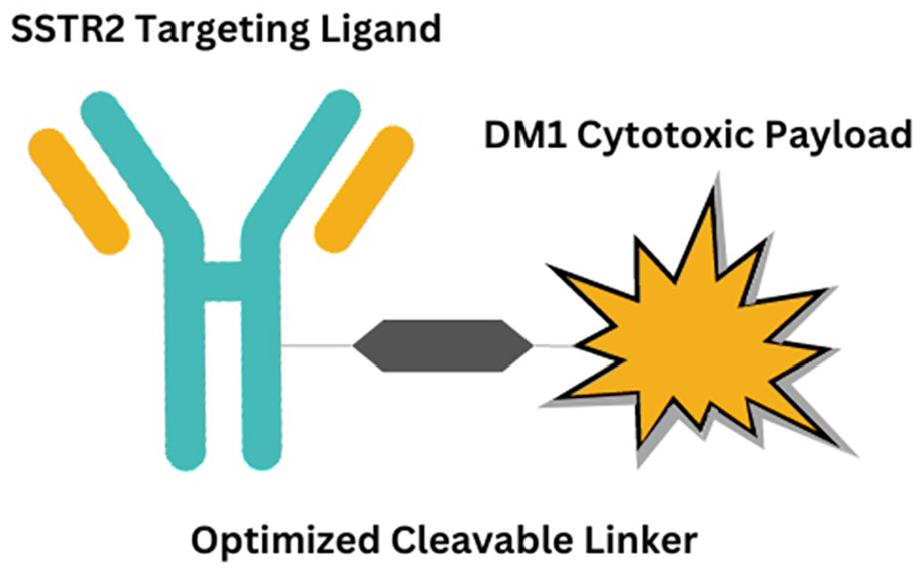
PEN-221 somatostatin analog (SSA)-DM1 conjugate structure.
Current Bispecific Antibodies in NECs
BI 764532 (DLL3 × CD3)
Delta-like ligand 3 (DLL3) is a type I transmembrane protein and a Notch receptor ligand. It plays an important role in the regulation of the Notch signaling pathway that impacts tumor growth, migration, and invasion. 16 It is diffusely expressed on the cell surface of small cell lung cancer (SCLC) and other NECs.17 -19 Previous results demonstrated it to be a valuable histological marker for the diagnosis of poorly differentiated NECs and distinguishes them from high-grade well-differentiated NETs. 20 Preclinical data showed that significant growth inhibition through the induction of intrinsic apoptosis was associated with DLL3 gene silencing. 21 Therefore, the high percentage of DLL3 expression in NEC highlighted it as a potential target for this subgroup of aggressive tumors. BI 764532 is a bispecific DLL3/CD3 IgG-like T-cell engager (TcE) which encourages concomitant binding to DLL3 on tumor cells and CD3 on T-cells leading to T-cell mediated antitumor cytolytic activity (Figure 3). 22 In preclinical studies, it achieved T-cell activation and lysis of DLL3-positive SCLC tumor cells In addition, in vivo studies conducted in a human T-cell engrafted xenograft model, the DLL3/CD3 bispecific Abs lead to an increase in T-cells infiltration into the tumor tissue, causing apoptosis of the tumor cells and complete tumor regression. 23 The first in human phase I study of BI 764532 monotherapy in patients with DLL3-positive SCLC and NECs (NCT04429087) was recently completed. 22 The results presented at the 2023 ASCO Annual meeting showed that BI 764532 at dose of 90 μg/kg elicited a partial response in 25% of the overall population (n = 71), and 19% of extrapulmonary NECs (n = 27). The median duration of response (DOR) has not been reached, with a disease control rate of 52% for all patients given a dose of 90 μg/kg or more. Dose limiting toxicities were reversible and occurred in only 5 of 107 patients treated on the study at all dose levels. Grade 3/4 CRS occurred in 2 patients; grade 3 confusional state and a grade 3 nervous system disorder occurred in 1 patient each. These results demonstrated that BI 764532 at dose of 90 μg/kg were well tolerated and was associated with durable responses in EP-NEC.

Structure and mechanism of action of BI 764532. 22
HPN 328 (DLL3 × CD3)
Another tri-specific, half-life extended DLL3-targeted T-cell engager is HPN 328 (Figure 4). 24 HPN 328 contains 3 important binding domains: anti-DLL3 for target engagement, anti-CD3 for T-cell engagement, and anti-albumin for half-life extension. 6 It demonstrates a cellular cytotoxicity against DLL3-expressing SCLC and extra-pulmonary-NEC. A phase I/IIA clinical trial (NCT04471727) that is currently testing HPN 328 for relapsed or refractory SCLC patients and any DLL3-expressing NECs has had astonishing preliminary results. 25 In 16 patients receiving an 8 dose-escalation, HPN328 was well tolerated and clinically active without grade 3 CRS, dose-limiting toxicities, or adverse events that would lead to discontinuation of the drug. Seven of 18 patients (39%) had any decrease in sum of target lesion diameters (5 SCLC, 1 extra-pulmonary-NEC, and 1 thymic atypical carcinoid). One patient with SCLC had partial response, 3/11 (27%) SCLC patients across all doses had > 30% decrease in sum of target lesion diameters, and 6 of 18 (33%) patients with best overall response of SD (4 SCLC, 1 extra-pulmonary-NEC, 1 thymic atypical carcinoid). The study is still ongoing and waiting for the final updated data.

Mechanism of action for HPN328.
PT217 (DLL3 × CD47)
PT217 is a BsAb that targets DLL3 and CD47 in patients with SCLC and other NECs (Figure 5).26,27 CD47 is a membrane protein that is widely distributed on the surface of various cells, including tumor cells. It acts as a signal indicating a message equivalent to “do not eat me,” acting as an inhibitor of phagocytosis through ligation of signal-regulatory protein alpha (SIRPα) expressed on phagocytes. Significantly, this leads to tyrosine phosphatase activation and inhibition of myosin accumulation at the submembrane assembly site of the phagocytic synapse. Therefore, inhibiting the expression of CD47 molecules in tumor cells, or even blocking the pathway between CD47 and SIRPα, can reactivate the phagocytosis of tumor cells by macrophages. PT217 targets both CD47 and DLL3 which is overexpressed in SCLC cells and NEC. 28 This blocks the interaction between CD47 and SIRPα and mediates potential antibody-dependent cytotoxicity of NK cells against tumor cells. Preclinical studies showed that PT 217 had an increased total selective binding when compared with anti-DLL3 monoclonal antibodies. In addition, results showed that PT217 stimulated antibody-dependent cellular phagocytosis of tumor cells by macrophages. 26 Currently, there is an ongoing phase I clinical trial (NCT05652686) to test the safety, tolerability, pharmacokinetics (PK), and preliminary efficacy of PT217 in patients with advanced refractory DLL3-expressing (SCLC), large-cell neuroendocrine cancer (LCNEC), neuroendocrine prostate cancer (NEPC), and gastroenteropancreatic neuroendocrine carcinoma (GEP-NEC) that have progressed after all available standard therapy.

Mechanism of action for PT 217.
Conclusions
Bispecific antibodies are an emerging modality for management of GEP-NENs including both well-differentiated NETs and high-grade NECs. The current early data regarding their tolerability, efficacy, and outcome are promising. Multiple BsAbs are in different phases of clinical trials but further studies are required to establish their long-term outcomes and relevant adverse events. Further phase II and III clinical trials are needed to reach better results for their implication and sequencing in the management of GEP-NENs.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Mai Elhawi: Writing the manuscript; Marcus Trybula: Edit the manuscript and the figures; Sylvia L Asa: editing the manuscript; Mohamed Elshawy: Writing the manuscript; Sakti Chakrabarti: Editing the manuscript; Eva Selfridge: Editing the manuscript; Amr Mohamed: Overview, writing and editing the manuscript.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.