
Abstract
Background:
Our previous research showed that Porphyromonas gingivalis (P. gingivalis) infection can activate the inflammatory signaling pathway and promotes the malignancy development of esophageal squamous cell carcinoma (ESCC). However, the prognostic significance of inflammatory response-related genes (IRRGs) in P. gingivalis-infected ESCC requires further elucidation. Hence, our study constructed a prognostic signature based on P. gingivalis and IRRGs to forecast the survival of patients with ESCC, which may provide insight into new treatment options for ESCC patients.
Methods:
Differentially expressed genes (DEGs) were identified in P.gingivalis-infected and P.gingivalis-uninfected ESCC cell by RNA sequencing. A risk model was constructed and validated using the The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) database by using univariate Cox regression analysis, LASSO, and the multivariate Cox regression analysis. Kaplan-Meier analysis was carried out to compare the overall survival (OS) between high-risk and low-risk groups. Single-sample gene set enrichment analysis was used to analyze the immune cell infiltration. The Genomics of Drug Sensitivity in Cancer database was used to predict drug sensitivity.
Results:
There were 365 DEGs between the P.gingivalis-infected and P.gingivalis-uninfected groups. Four genes including DKK1, ESRRB, EREG, and RELN were identified to construct the prognostic risk model (P = .012, C-index = 0.73). In both the training and validation sets, patients had a considerably shorter OS in the high-risk group than those in the low-risk group (P < .05). A nomogram was established using the risk score, gender, and N stage which could effectively forecast the prognosis of patients (P = .016, C-index = 0.66). The high-risk group displayed lower immune infiltrating cells, such as activated dendritic cells, type 2 T helper cells, and neutrophils (P < .05). A total of 41 drugs, including dactinomycin, luminespib, and sepantronium bromide, had a significant difference in IC50 between the 2 subgroups.
Conclusion:
We demonstrated the potential of a novel signature constructed from 4 P. gingivalis-related IRRGs for prognostic prediction in ESCC patients.
Keywords
Introduction
Esophageal cancer (EC) is a malignant tumor that has a significant incidence and fatality rate. In 2020, there were 604 100 new cases and 544 076 deaths of EC worldwide. 1 This ranks EC as the seventh most common cancer by incidence and the sixth deadliest malignancy. China, in particular, bears a high burden of EC, with more than 90% of cases being esophageal squamous cell carcinoma (ESCC). 2 In 2020, China reported 324 000 new cases and 301 000 deaths from EC, accounting for ~55% of the global figures.1,3 Despite advancements in multimodal therapies such as surgical procedures, chemotherapy, radiotherapy, and targeted therapy, the overall 5-year survival rate for EC is still less than 20%. 4 Hence, it is crucial to identify novel prognostic signatures to assess individualized survival risk for patients with ESCC.
Mounting evidence suggests that the microbiome of the esophagus, along with inflammation and their interaction, play a crucial role in promoting EC. 5 It is estimated that 25% of all malignant tumor cases worldwide can be attributed to long-term infection and inflammation. 6 Porphyromonas gingivalis (P. gingivalis), a significant pathogenic bacterium associated with local immune inflammatory responses in long-term periodontitis, has garnered attention in recent years due to its close association with various malignant tumors.7,8 Porphyromonas gingivalis has been found to be closely related to ESCC in recent years. In 2016, a study reported found that the infection rate of P. gingivalis in ESCC tissues (61%, 71%) were significantly higher than adjacent normal tissue (12%, 12%) and normal esophageal mucosal tissue (0%, 1%) by immunohistochemistry (IHC) and polymerase chain reaction (PCR). In addition, the mean survival time of P. gingivalis-positive patients was significantly lower than that of negative patients. 9 Another study discovered that serum P. gingivalis IgG and IgA antibody levels in ESCC patients were significantly higher than those in esophagitis patients and healthy control group (P < .01). And high levels of P. gingivalis IgG or IgA antibodies are associated with poor prognosis in ESCC patients. 10 Besides, there is enrichment of P. gingivalis in high-grade dysplasia and early ESCC, and overabundance of P. gingivalis was positively associated with invasion depth, postendoscopic submucosal dissection stricture and local recurrence. 11 Notably, patients with ESCC who were infected with P. gingivalis had poor chemotherapy responses and a significantly shortened 5-year survival rate following surgical intervention.12,13 In conclusion, the above researches showed that P. gingivalis had a significant influence on the development and progression of ESCC, as well as its impact on the patient prognosis. Long-term inflammation is a key player in oncogenesis and tumor progression. Long-term inflammation induced by microbial infection significantly elevates the possibility of carcinogenesis by way of activating inflammatory signaling pathways and cytokines, stimulating cell proliferation, and inhibiting apoptosis. 14 There is clear evidence that long-term colonization of Helicobacter pylori, human papillomavirus, and hepatitis B virus can accelerate the progression of stomach carcinoma, cervical carcinoma, and hepatocellular carcinoma by inducing long-term inflammation. 15 Wang et al 16 indicated that nuclear factor (NF)-kappa BP65 promotes invasion and metastasis of ESCC by regulating MMP9 and epithelial-mesenchymal transition (EMT). Besides, up-regulation of interleukin (IL)-6/STAT3 signaling pathway promotes the occurrence of EC. 17 Interleukin-6 is a cytokine that binds to gp130 through its receptor IL-6Rα, triggering the downstream pathway to activate important molecules such as SHP2, Ras-MAPK, STAT1, and STAT3. 18 These pathways activate the ability of tumor cells to survive in the inflammatory environment and inhibit the effects of immunotherapy. Interleukin-6 can drive the proliferation of myeloid-derived suppressor cells (MDSCs), and the activation of STAT3 leads to the production of antiapoptotic molecules, thus causing tumorigenesis. 19 P. gingivalis has been proven to induce inflammation and promote the malignant progression of ESCC by activating STAT3, GSK3β, NF-κβ, and tumor growth factor (TGF)-β signaling pathways.8,20 In addition, the intratumoral P. gingivalis promotes the progression of pancreatic cancer by enhancing the secretion of neutrophilic chemokines and neutrophil elastase. 21 The fibrinogen/albumin ratio, neutrophil/lymphocyte ratio, and platelet/lymphocyte ratio, as markers of inflammation in circulating blood, have been shown to be associated with the prognosis of EC.22,23 Previous studies have reported models based on inflammatory responses for prognostic predictions in EC. 24 This finding suggested that inflammation has an impact on cancer prognosis and can serve as a promising therapeutic target. However, the role and prognostic significance of inflammatory response-related genes (IRRGs) in P. gingivalis-infected ESCC remain unclear.
We first constructed an IRRG prognostic signature by using The Cancer Genome Atlas (TCGA)-ESCC data set and the RNA-seq data of P. gingivalis-infected ESCC cells in this study. Then we validated its prognostic value using the GSE53622 data set. Thereafter, a nomogram was constructed to predict the survival of patients with ESCC. Simultaneously, we explored the relationship between IRRGs and immune function, genetic mutation analysis, immunotherapy, and drug susceptibility. Our findings have the potential to improve comprehension of the prognostic mechanisms and provide novel and valuable biomarkers for treating patients of ESCC. The flowchart of this study is exhibited in Figure 1.

Flowchart of the present study.
Materials and Methods
Cell culture, bacteria strain, and cell infection
The ESCC cell line Kyse-140 (BNCC351870, BeNa Culture Collection, China) was cultured at 37°C with 5% CO2 in RPMI-1640 medium (PM150110, Procell, China) supplemented with 10% fetal bovine serum (164210-500, Procell, China). P. gingivalis ATCC 33277 was donated by the University of Louisville and was cultured at 37°C under anaerobic conditions consisting of 85% N2, 10% H2, and 5% CO2 in Brain Heart Infusion (BHI) broth medium (237500, Becton Dickinson, USA) containing 0.1% Hemin and 0.1% Vitmin K1. The optical density of bacterial solution was measured at 600 nm by spectrophotometer with a value of 1.0 corresponding to 1 × 109 CFU/ml. Then centrifuge the bacterial solution at 12 000 g for 5 minutes, and remove the supernatant. Resuspend the P. gingivalis bacterial precipitate in 1 ml phosphate-buffered saline (PBS) and add it to Kyse-140 cell culture medium. Finally, cells were inoculated with P. gingivalis at a multiplicity of infection (MOI) of 20 for 24 hours at 37°C in 5% CO2.
Data sources
In this study, we performed transcriptome sequencing of Kyse-140 cells between P. gingivalis-infected and P. gingivalis-uninfected in the GENEWIZ Biotechnology Corporation. The TCGA-ESCC data set containing 80 tumor specimens and 11 normal specimens with clinical information and survival information (Table 1) available was sourced from the TCGA database (https://www.cancer.gov/ccg/). Moreover, we used the GSE53622 data set was achieved from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/), containing 60 tumor samples and 60 control samples, as an external validation data set (Table 1). In addition, 3074 IRRGs were gathered from the GeneCards online database according to a score threshold >5. Furthermore, the IMvigor210 data set containing 298 cohort data of immunotherapy for bladder urothelial carcinoma (BLCA) was obtained from the IMvigor210 database.
Clinicopathological characteristics of ESCC patients in TCGA and GEO cohort.

Acquisition analysis and enrichment analysis of crossover genes
Differentially expressed genes (DEGs) between the P. gingivalis-infected and P. gingivalis-uninfected group were identified using the DESeq2 (v1.36.0) R package (P < .05). 25 Moreover, DEGs between ESCC and normal samples in the TCGA-ESCC data set were identified by the same method. To visualize the DEGs, we generated heat maps and volcano maps for the P. gingivalis-infected versus P. gingivalis-uninfected groups, and tumor versus normal groups were plotted by the heatmap (v0.7.7) and ggplot2 (v3.3.0) R packages, respectively. 26 Furthermore, we performed an intersection analysis to identify genes that were common between the DEGs in the self-sequencing data, DEGs in the TCGA-ESCC data set, and the set of IRRGs. In addition, to acquire understanding of the biological functions and signaling pathways associated with these crossover genes, we performed gene ontology (GO) functional enrichment analysis using the clusterProfiler (v3.8.1) R package with a significance threshold of P < .05. 27
Construction and verification of the prognostic model
The Cancer Genome Atlas-ESCC data set was classified into a training set and an internal validation set using a 1:1 ratio, and the GSE53622 served as an external validation set. First, the univariate Cox regression analysis was performed on the above crossover genes to screen the candidate genes (P < .2). 28 Then, the LASSO algorithm was executed on candidate genes acquired through the univariate Cox regression analysis (family = Cox). When constructing the model, the genes with coefficients of the variables retained in the model with the smallest Partial-likelihood deviance were selected; these genes were used to construct the multivariate cox model, and when constructing the multivariate cox model, the models were scored using the STEPWISE algorithm. The genes retained in the highest scoring model were selected as the key genes, and the highest scoring model was the final risk scoring model used. A stepwise approach was then performed to optimize the model and identify prognostic genes. This process resulted in the creation of a risk model of prognostic genes.
To divide samples into high- and low-risk groups, the risk score for each sample was computed using the expression levels of these prognostic genes. The risk score was computed according to the median value of risk as follows: (risk score =
Independent prognostic analysis
First, the univariate Cox regression analysis was implemented on the risk score and clinical features such as tumor pathologic T-M-N, stage, gender, and age. Then, the variables obtained by the univariate Cox regression analysis were used to create the multivariate Cox model. The proportional hazards (PH) hypothesis test was implemented to verify the validity of the Cox model. Furthermore, according to the above prognostic model, a nomogram for forecasting survival rates of patients with ESCC (1-, 2-, and 3 year survival rates) was created. Finally, we drew a calibration curve to verify the effectiveness of the above model.
Differential expression analysis and enrichment analysis
Differentially expressed genes between the 2 risk subgroups were acquired by the DESeq2 (v1.36.0) R package (P < .05 and|log2FC| > 2). 25 To explore the associated biological functions and signaling pathways of the above DEGs, the GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analyses were performed using the clusterProfiler (v3.8.1) R software package (adjusted P < .05). 27
Genetic mutation analysis
The mutation analysis of prognostic genes in the TCGA-ESCC data set was conducted using the maftools (v2.12.0) R package. 29 The mutual exclusion and co-occurrence of the most common top 25 mutant genes were analyzed using the CoMEt algorithm.
Analyses of immune characteristics
To further explore the immune infiltration environment of ESCC, the CIBERSORT algorithm was performed on the samples in the TCGA-ESCC data set. This algorithm estimates the abundance of 22 different immune cell types, allowing for a comparison of immune cell populations between the 2 risk subgroups. In addition, in the TCGA-ESCC data set, the differential immune cells between the 2 risk subgroups were computed and compared by ssGSEA algorithm and Wilcoxon test method, respectively. The risk score was computed using the expression of prognostic genes in the IMvigor210 data set. The best cut-off threshold for risk score classification was calculated using the surv-cutpoint function, resulting in the classification of samples into high-risk and low-risk groups. Finally, we conducted a K-M survival analysis between the 2 risk subgroups to evaluate the prognostic value of the risk score in predicting patient survival.
Drug sensitivity prediction
The IC50 of chemical drugs for patients with ESCC was forecasted according to the cell line expression profile and gene expression profile in GDSC online database (https://www.cancerrxgene.org/). The Oncopredict tool was used to compute the IC50 of common chemotherapy and molecular-targeted drugs in patients with ESCC. Besides, the Wilcoxon test method was used to analyze the IC50 of chemical drugs with remarkable differences in sensitivity between the 2 risk subgroups.
Statistical analysis
All the analyses were implemented in the R (v4.2.0) package and the related public databases. The Wilcoxon rank-sum test was used for comparison between 2 groups. Cox and Lasso regression analysis were used to reduce the dimension of model-related risk IRRGs and construct a polygenic prognostic risk model. Kaplan-Meier survival curves are plotted and log-rank test was conducted between different subgroups. Receiver operating characteristic curves were constructed for predicting survival at 1, 2, and 3 years. A 2-sided P < .05 was considered statistically significant.
Results
Identification of IRRGs DEGs of P. gingivalis-infected ESCC
In our study, we identified 365 DEGs in the self-sequencing data set, including 217 up-regulated and 148 down-regulated genes between the P. gingivalis-infected group and the P. gingivalis-uninfected group (Figure 2A, Supplementary Table 1). Moreover, within the TCGA-ESCC data set, we found 9817 DEGs between the tumor and normal samples, of which 4989 were up-regulated and 4833 were down-regulated (Figure 2B, Supplementary Table 2). The respective expression of these DEGs was shown in heat maps (Figure 2C and D). In addition, 3074 IRRGs were gathered from the GeneCards online database according to a score threshold >5. According to the intersection of DEGs in the self-sequencing data set, DEGs in the TCGA-ESCC data set, and the set of IRRGs, 66 crossover genes were identified using a Venn map (Figure 2E). According to the enrichment analysis, crossover genes were primarily linked to biological processes like “epidermis development,” “skin development,” and “cytokine-mediated signaling pathway” in GO terms (Figure 2F, Supplementary Table 3).

Identification of candidate IRRGs. (A) the volcano plot of DEGs between P. gingivalis-infected and P. gingivalis-uninfected ESCC cells. (B) The volcano plot of DEGs in the TCGA-ESCC data set. (C) The clustering heat map of the DEGs between P. gingivalis-infected and P. gingivalis-uninfected ESCC cells. (D) The clustering heat map of the DEGs in the TCGA-ESCC data set. (E) The Venn diagram to identify DEGs. (F) Representative results of GO analyses in TCGA.
Construction of risk models
There were 7 candidate genes including TNFRSF12A, REEP1, DKK1, FOXN1, ESRRB, EREG, and RELN (Figure 3A). A total of 6 candidate prognostic genes (TNFRSF12A, DKK1, FOXN1, ESRRB, EREG, and RELN) were identified by the LASSO algorithm (Figure 3B). According to the multivariate Cox regression analysis, 4 prognostic genes including DKK1, ESRRB, EREG, and RELN were screened out. Notably, the expressions of DKK1, ESRRB, and EREG negatively related to the prognosis, while RELN exhibited the opposite pattern (Figure 3C). A prognostic signature for patients with ESCC was constructed based on the expressions of these 4 prognostic genes and their Cox regression coefficient (β). The risk score was computed as follows: Riskscore = (–0.33617 × DKK1 expression) + (–0.57111 × ESRRB expression) + (–0.27303 × EREG expression) + (0.319031 × RELN expression). The prognostic model’s performance was assessed, demonstrating a C-index of 0.73, which was substantially higher than the C-index of the individual genes (Figure 3D). This indicates that the model has a strong predictive ability for patient prognosis.

Screening of prognostic genes and construction of prognostic models. (A) The prognostic genes identified by univariate analysis in the TCGA cohort. (B) LASSO coefficient profiles and selection of the number of genes by LASSO analysis. (C) The 4 target genes identified by multivariate Cox regression analysis. (D) The C-index value of the prognostic model and prognostic gene.
Validation of the prognostic risk model
The risk scores of high- and low-risk groups were significantly different across these 3 data sets, (P < .05). Moreover, the number of deaths increased in all 3 data sets with an increased in the risk score. The expression of prognostic genes in the 3 data sets was displayed in the heat maps (Figure 4A to C). The expression of DKK1, ESRRB, and EREG were elevated in the low-risk group, whereas RELN was decreased. The accuracy of predicting 1-, 2-, and 3-year survival prognosis was evaluated using the time ROC curve and area under the curve (AUC) value. The results showed that the AUCs of the ROC curves were all exceeded 0.6 in both the training set and external validation set, indicating that the created risk model could effectively predicted the survival rates of patients with ESCC (Figure 4D to F). Next, we plotted a confusion matrix for risk grouping versus survival status, which showed an accuracy of 0.5823, a sensitivity of 0.7750, and a specificity of 0.3846 (Supplementary Figure 1). Furthermore, K-M analysis indicated that there was a remarkable difference in the prognosis of patients with ESCC between 2 risk subgroups, with the high-risk group having significantly worse outcomes than those in the low-risk group in all 3 data sets (P < .05) (Figure 5A to C). Finally, there were obvious differences in the patterns of gene expression among the 2 risk subgroups in the 3 gene sets, supporting the notion that the prognostic genes could effectively distinguish between these 2 risk subgroups (Figure 5D to F).

Validation and evaluation of risk models in both the TCGA and GEO cohorts. (A to C) The distribution of the risk scores (top), survival status (middle), and expression heat map of the 4 IRRGs (bottom). (D to F) The time ROC curve for OS.

High and low risk group survival analysis and PCA validation. (A to C) Kaplan-Meier curves for OS in the high- and low-risk groups. (D to F) PCA plot in the high- and low-risk groups.
The nomogram for overall survival prediction
We found that risk score, pathologic N, and gender were all connected with the survival and prognosis of patients with ESCC (Figure 6A). Meanwhile, the model’s overall performance was assessed, yielding a P value of .0165 and a C-index of 0.66 (Figure 6B), indicating a significant overall association with survival outcomes and a moderate level of predictive accuracy. The PH hypothesis test showed that neither the individual covariates nor the global test were statistically significant (P > .05). Therefore, the Cox model conformed to the PH assumption (Figure 6C). Moreover, a nomogram for survival forecasting in patients with ESCC (1, 2, and 3 years) was created based on risk score, pathologic N, and gender. The nomogram estimates the overall survival (OS) by adding up the points assigned to each parameter based on the provided scale. The risk score was found to have a substantial contribution to prognosis (Figure 6D). Finally, a calibration curve was plotted to assess the effectiveness of the nomogram. The curve indicated that the nomogram model had favorable prediction ability (Figure 6E).

The nomogram constructed in the TCGA-ESCC cohort. (A) Forest plots for univariate cox regression analysis. (B) Forest plots of independent prognostic model. (C) PH hypothesis test. (D) The nomogram for predicting OS. (E) Calibration curves for the nomogram.
Functional enrichment analysis of DEGs in high- and low-risk groups
There were 205 DEGs between the 2 risk subgroups (Figure 7A, Supplementary Table 4). The expression of DEGs is shown in the heat map (Figure 7B). According to the GO analysis, DEGs between the 2 risk subgroups are mainly involved in the xenobiotic metabolic process, cellular response to xenobiotic stimulus, and retinoid metabolic process in biological processes. For cellular components, DEGs were enriched in components like the integral component of the synaptic membrane, an intrinsic component of the synaptic membrane, and an integral component of the postsynaptic membrane. For molecular functions, the DEGs were linked to activities such as carboxylic ester hydrolase activity, monocarboxylic acid binging, and glutathione transferase activity. The KEGG pathways enrichment analysis results indicated that the DEGs between the 2 risk subgroups were primarily participated in chemical carcinogenesis-receptor activation, metabolism of xenobiotics by cytochrome P450, drug metabolism-cytochrome P450, chemical carcinogenesis-DNA adducts, and drug metabolism-other enzymes (Figure 7C to F, Supplementary Table 5-6).

GO and KEGG analyses using the DEGs between the high- and low-risk groups. (A) Volcano plots of the DEGs. (B) The heat map of DEGs. (C and D) GO enrichment analysis based on DEGs. (E and F) KEGG enrichment analysis based on DEGs.
Genetic mutation analysis in high- and low-risk groups
From the TCGA-ESCC data set, the proportion of missense mutations was the highest among the mutation types, and the top 10 mutated genes were TP53, TTN, KMT2D, CSMD3, NFE2L2, NOTCH1, MUC16, FLG, PIK3CA, and DNAH5 (Figure 8A). It was found that TP53 had the highest mutation rate amongst all genes in both subgroups. In the high-risk group, mutations in NFE2L2, CSMD3, and MUC16 were most prevalent. In contrast, in the low-risk group, DNAH5, KMT2D, and MUC17 had a higher mutation rate (Figure 8B). Notably, KMT2D and TP53 exhibited mutual exclusivity, meaning these 2 genes were less likely to co-occur in the same patient, in the low-risk group. In addition, certain genes such as FAT3 and TTN, COCH, and SACS, exhibited obvious co-occurrence, suggesting that mutations in these gene pairs were more likely to appear together in the same patient, in the low-risk group. Furthermore, no genes showed obvious mutual exclusion in the high-risk group, but there was an obvious co-occurrence between MUH13 and FLG, as well as between MUH13 and ELAPOR2 (Figure 8C and D).

Genetic mutation in high- and low-risk groups of patients with ESCC. (A) An overview of genetic mutations. (B) The waterfall plot of genetic mutation features. (C) Co-occurrence and mutual exclusion of mutated genes in the low-risk group. (D) Co-occurrence and mutual exclusion of mutated genes in the high-risk group.
Analyses of immune characteristics in high- and low-risk groups
The abundance of immune cells in the 2 subgroups was displayed in Figure 9A. The results showed remarkable differences in 3 immune cells (activated dendritic cells [DCs], type 2 T helper cells, and neutrophils) among the 2 risk subgroups (Figure 9B). The composition of these immune cells was found to be more abundant in the low-risk group compared with the high-risk group. The IMvigor210 data set was used to analyze the relationship and predictive power of risk score for programmed death ligand 1 (PD-L1) immune efficacy. There was little difference in the number of patients with stable disease between the 2 subgroups; however, the number of patients with ESCC with partial remission and complete remission in the high-risk group was significantly higher (Figure 9 C). We also found a statistical significance in survival prognosis between the 2 subgroups, and the lower risk scores had poorer OS (P = 0.029) (Figure 9D).
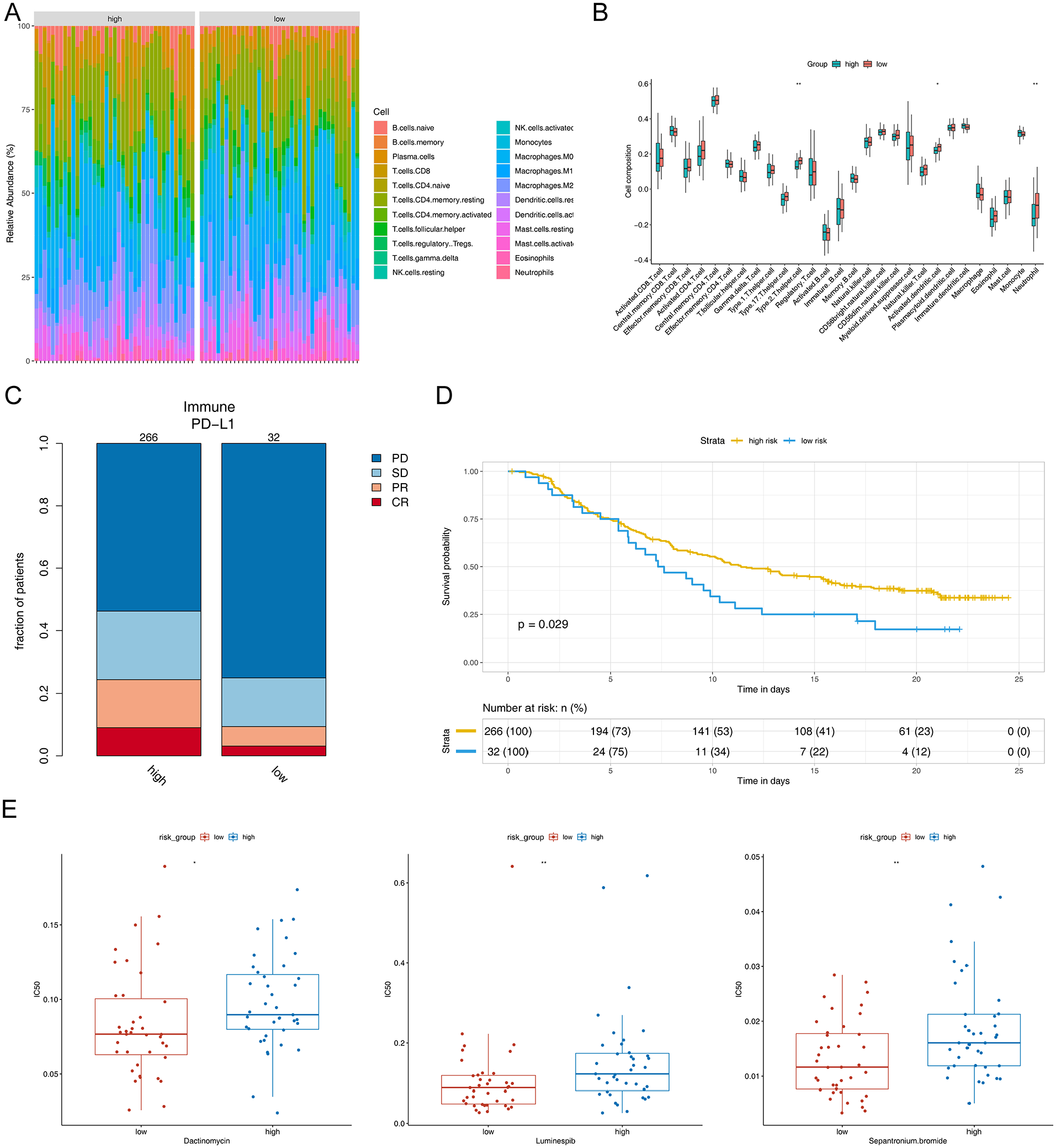
Immune characterization correlation analysis and drug susceptibility prediction. (A) Immune cell abundance map of each sample. (B) The boxplots of immune infiltration analysis. (C) Comparison of efficacy of immunotherapy between the 2 groups. (D) Kaplan-Meier curves of the OS in external validation data sets. (E) Sensitivity to dactinomycin, luminespib, and sepantronium bromide shown in the box plot.
Drug sensitivity prediction in high- and low-risk groups
The analysis identified 41 drugs with a statistical difference in IC50 values between the 2 risk subgroups. Notably, drugs such as sepantronium bromide, dactinomycin, and luminespib all exhibited lower IC50 values in the low-risk group (P < .001) (Figure 9E). On the whole, low-risk groups were more susceptible to sepantronium bromide, dactinomycin, and luminespib which means these patients may have a more favorable response to these drugs, potentially leading to improved treatment outcomes.
Discussion
Esophageal cancer is a malignant tumor with high mortality. With the advancement of medical technology, a variety of treatment methods have been improved for ESCC. However, we often cannot accurately forecast the effectiveness of ESCC treatment because of lack reliable biomarkers. Numerous studies have indicated that P. gingivalis and inflammation response play a crucial role in the pathophysiology and malignant progression of ESCC, which is associated with patient prognosis.5,9 However, the role of P. gingivalis and IRRGs as prognostic predictors for ESCC remains to be elucidated. Thus, it is particularly critical to explore accurate biomarkers for predicting prognosis of P. gingivalis-infected ESCC patients. In this study, we first constructed and verified a prognostic gene signature composed of P. gingivalis and inflammation-related DEGs, namely DKK1, ESRRB, EREG, and RELN. This signature serves as an independent risk factor to forecast the prognosis of patients with ESCC. Furthermore, we established straightforward and user-friendly prognostic nomogram model to assist in forecasting the 1-, 2-, and 3-year OS, which may contribute to the clinical treatment of patients with ESCC. Although the prognostic model established in this study through the cell models in vitro is not representative of the microenvironment in vivo, previous studies have confirmed that P. gingivalis infection cells in vitro can affect the tumor microenvironment and promote the development of tumors.12,13,30
Previous research has established the roles of these 4 prognostic genes in cancer progression. DKK1 encodes a secreted protein that is an antagonist of the Wnt/b-catenin signaling pathway, which is participate in tumor progression and is overexpressed in numerous human tumors.31,32 DKK1 is closely linked to prognosis and has been identified as a possible diagnostic and prognostic indicator for ESCC.33,34 Recent research has evidenced that DKK1 stimulates cancer cell proliferation by activating the PI3K-AKT pathway. 35 Furthermore, DKK1 has been demonstrated to sustain the tumor stemness of EC cells through ALDH1A1/SOX2 axis. 36 ESRRB, a nuclear transcription factor, controls self-renewal and pluripotency of embryonic stem cells. 37 Studies have discovered that ESRRB is associated with tumor progression of breast and prostate cancers.38,39 In addition, ESRRB accelerates the growth of carcinoma cells in vitro and in vivo by repressing the TGF-β pathway by transactivating SMAD7. 40 Our study represents the first discovery of ESRRB as a prognostic biomarker for ESCC, although further studies are warranted to elucidate its prognostic significance and its underlying mechanisms in patients with ESCC. EREG, a ligand for the estimated glomerular filtration rate (EGFR) family, contributes to processes including angiogenesis, vascular remodeling, cell proliferation, and inflammation. 41 Previous studies have noted that EREG is typically low or absent in most human tissues but is found to be over-expressed in various tumors. 42 EREG has been involved in promoting the invasion of EC, formation of spheres, reorganization of actin, and lung metastasis by activating FAK and Src. 43 RELN is a well-known large extracellular matrix glycoprotein expressed primarily in brain development, where it regulates neuronal migration, adhesion, and positioning. 44 However, recent investigations have shown abnormal expression patterns of RELN in various cancers. 45 RELN is a critical negative regulator during TGF-β1-induced cell migration of Kyse-30 cells and is inhibited through the TGF-β pathway via Snail regulation at the transcriptional level. 46 Our study found high expressions of DKK1 and EREG and low expressions of ESRRB and RELN in patients with ESCC, which was consistent with the above studies. Therefore, it is speculated that DKK1, ESRRB, EREG, and RELN serve as key regulatory factors in the carcinogenic process of P. gingivalis.
Then, we would like to further analyze the underlying mechanism of IRRGs in P. gingivalis-infected ESCC. The findings from KEGG and GO analyses indicated that DEGs primarily participated in processes such as the chemical carcinogenesis-receptor activation, metabolism of xenobiotics by cytochrome P450, drug metabolism-cytochrome P450, chemical carcinogenesis-DNA adducts. For instance, CYP2C9 participates in the metabolism of tumor drugs and exogenous carcinogens, which suppresses the invasion and migration of ESCC by reducing the levels of HDAC. 47 Similarly, CYP1A1 is involved in the oxidative conversion of xenobiotics, whereas its metabolic reactions may unintentionally result in the production of highly reactive compounds, which can form DNA adducts, thereby promoting mutagenesis and carcinogenesis. 48 The findings suggested that P. gingivalis could affect the changes in the ESCC metabolic pathway by regulating IRRGs.
Numerous pieces of evidence have demonstrated that the tumor immune microenvironment plays a crucial role in carcinogenesis. The results showed that the immune state was significantly different between the low- and high-risk patients with ESCC. Notably, immune-infiltrating cells were more abundant in the low-risk group, with heightened levels of activated DCs, type 2 T helper cells, and neutrophils. This suggests that immune regulation may be inhibited in the high-risk group, contributing to a worse prognosis in patients with ESCC. Dendritic cells are specialized antigen-presenting immune cells, which have a crucial function in the immune response against cancer. 49 Furthermore, research has indicated that increases in the number of DCs can improve immune function and extend the survival time of patients with EC. 50 Dendritic cells can kill tumor cells by stimulating the proliferation of T cells, ultimately inhibiting tumor cell proliferation and metastasis. 51 Type 2 T helper cells have an important influence on type-2 immune responses and enhance the production of antibody against extracellular tissue. 52 Research has shown the infiltration level of type 2 T helper cells is positively correlated with OS in patients with ESCC without chemotherapy after surgery, with higher infiltration levels of these cells associated with longer OS. 53 However, in the context of long-term inflammation, type 2 T helper cells have been associated with tumor promotion by provoking humoral immune responses and interfering with the recruitment and activation of cytotoxic T lymphocytes in tumor. 54 Neutrophils, often considered the first line of defense against inflammations and infections, 55 can adopt differential states of activation and differentiation in various tumor contexts. They can polarize into either an anti-cancerous (N1) type or a cancer-promoting (N2) type. 56 A recent research identified that MPO + neutrophils infiltrating the tumor were associated with a positive prognosis for ESCC. 57 When TGF-β is not present, the N1 neutrophils secrete more immune activating cytokines and chemokines, with lower levels of arginase, and exhibit an improved capacity to eliminate cancerous cells in vitro and in vivo. 58 In summary, the immune cells above are potentially involved in the regulation of immune response in EC development and are thought to be beneficial for survival, consistent with the observations of our current study. Notably, our research identified a decrease in the proportion of type 2 T helper cells, activated DCs, and neutrophils in high-risk patients with P. gingivalis infection. Therefore, it is speculated that P. gingivalis may weaken the antitumor immune response of the body by inhibiting the above immune cell infiltration. This could potentially be a significant factor contributing to the unfavorable prognosis of high-risk patients.
Finally, we further analyzed the drug sensitivity for ESCC according to the GDSC database. We discovered that the low-risk group was susceptible to chemotherapy drugs, for example, sepantronium bromide, dactinomycin, and luminespib. Sepantronium bromide (YM155), a survivin suppressant, has been demonstrated to enhance the radiation sensitivity of ESCC cells by suppressing radiation-induced senescence and promoting apoptosis. 59 In addition, YM155 improves radiosensitization by disrupting the G2 checkpoint and inhibiting homologous recombination repair in ESCC. 60 Luminespib, an HSP90 inhibitor, exhibits a strong antiproliferative effect for ESCC, hinting at it may become a new choice for the treatment of ESCC. 61 Therefore, the results of our study may hold promise in guiding decisions related to immunotherapy and chemotherapy, with the potential to inform clinical treatment strategies for patients with ESCC.
Our research has some limitations. First, for data selection bias, matching and sensitivity analyses can be further used in the future to reduce possible selection bias in the database. Then by comparing the results with external validation, the impact of selection bias on the findings can then be assessed and the generalizability, and reliability of the results can be improved. Second, our study was retrospectively analyzed, and the outcomes were only verified using the TGCA and GEO data sets, which requires more prospective studies and a large cohort of clinical tissue samples to validate the identified signatures and assess their relevance to immunity, prognosis, and resistance. In the future, our team keep on to explore this field. Finally, though P. gingivalis is related to the occurrence and development of ESCC, and the infection rate of P. gingivalis is about 60% due to individual differences and the influence of tumor microenvironment on micro-organisms. Hence, we established the prognostic model only for ESCC patients with P. gingivalis infection.
Although there are some limitations in our study, this does not affect the value of this study. First, the effect of IRRGs on the prognosis of P. gingivalis-infected ESCC patients has never been reported. Second, the predictive model is beneficial for more accurately predicting the prognosis of P. gingivalis-infected ESCC patients and improving the survival rate of patients. In the future, we can conduct more detailed microbiome analysis for patients with different subtypes of ESCC, as well as more in-depth studies combined with clinical data to verify the association between oral microbes and ESCC. In brief, our study not only reveals their role in ESCC but also lays the foundation for the early diagnosis and precision medicine of tumor.
Conclusions
In conclusion, we successfully developed a new predictive model for patients with ESCC, leveraging 4 IRGGs with prognostic significance. Moreover, our research also assessed the potential of the risk model to predict infiltration of immune cells and sensitivity to chemotherapy, thereby enhancing its clinical utility and providing potential biomarkers for clinical therapeutic strategies.
Supplemental Material
sj-csv-3-onc-10.1177_11795549241275666 – Supplemental material for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma
Supplemental material, sj-csv-3-onc-10.1177_11795549241275666 for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma by Jinyu Kong, Yiwen Liu, Jian Wang, Mengfan Qian, Wei Sun and Ling Xing in Clinical Medicine Insights: Oncology
Supplemental Material
sj-csv-4-onc-10.1177_11795549241275666 – Supplemental material for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma
Supplemental material, sj-csv-4-onc-10.1177_11795549241275666 for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma by Jinyu Kong, Yiwen Liu, Jian Wang, Mengfan Qian, Wei Sun and Ling Xing in Clinical Medicine Insights: Oncology
Supplemental Material
sj-csv-5-onc-10.1177_11795549241275666 – Supplemental material for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma
Supplemental material, sj-csv-5-onc-10.1177_11795549241275666 for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma by Jinyu Kong, Yiwen Liu, Jian Wang, Mengfan Qian, Wei Sun and Ling Xing in Clinical Medicine Insights: Oncology
Supplemental Material
sj-csv-6-onc-10.1177_11795549241275666 – Supplemental material for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma
Supplemental material, sj-csv-6-onc-10.1177_11795549241275666 for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma by Jinyu Kong, Yiwen Liu, Jian Wang, Mengfan Qian, Wei Sun and Ling Xing in Clinical Medicine Insights: Oncology
Supplemental Material
sj-csv-7-onc-10.1177_11795549241275666 – Supplemental material for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma
Supplemental material, sj-csv-7-onc-10.1177_11795549241275666 for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma by Jinyu Kong, Yiwen Liu, Jian Wang, Mengfan Qian, Wei Sun and Ling Xing in Clinical Medicine Insights: Oncology
Supplemental Material
sj-csv-8-onc-10.1177_11795549241275666 – Supplemental material for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma
Supplemental material, sj-csv-8-onc-10.1177_11795549241275666 for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma by Jinyu Kong, Yiwen Liu, Jian Wang, Mengfan Qian, Wei Sun and Ling Xing in Clinical Medicine Insights: Oncology
Supplemental Material
sj-docx-1-onc-10.1177_11795549241275666 – Supplemental material for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma
Supplemental material, sj-docx-1-onc-10.1177_11795549241275666 for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma by Jinyu Kong, Yiwen Liu, Jian Wang, Mengfan Qian, Wei Sun and Ling Xing in Clinical Medicine Insights: Oncology
Supplemental Material
sj-tif-2-onc-10.1177_11795549241275666 – Supplemental material for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma
Supplemental material, sj-tif-2-onc-10.1177_11795549241275666 for A Novel Porphyromonas gingivalis Infection-Related Inflammatory Response-Related Genes Signature Predicts the Prognosis of Esophageal Squamous Cell Carcinoma by Jinyu Kong, Yiwen Liu, Jian Wang, Mengfan Qian, Wei Sun and Ling Xing in Clinical Medicine Insights: Oncology
Footnotes
Acknowledgements
The author would like to thank Professor. Lamont RJ, and Professor Wang HZ at the University of Louisville for generously providing us with P. gingivalis strain ATCC 33277.
Author Contributions
JK wrote and revised the article. LX conceived and revised the article. JK and MQ contributed to the experimental studies. YL, JW, and WS revised the article and edited part of the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Medical Science and Technology Project of Henan Province (grant nos. SBGJ202103100 and SBGJ202103099); and the Science and Technology Projects of Luoyang (grant no. 2302010Y).
Availability of Data and Materials
The data sets involved in the study were all obtained from the TCGA database (https://www.cancer.gov/ccg/) and the GEO database (![]() ). All fastq raw sequence read data have been uploaded to the NCBI Sequence Read Archive (SRA) under accession number PRJNA1047328.
). All fastq raw sequence read data have been uploaded to the NCBI Sequence Read Archive (SRA) under accession number PRJNA1047328.
Consent for Publication
Not applicable
Ethics Approval and Consent to Participate
Not applicable
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.