
Abstract
Parathyroid carcinoma (PC) is an exceptionally rare endocrine malignancy characterized by severe hypercalcemia and high recurrence rates. We present the case of a 34-year-old male with chronic kidney disease who developed pathological fractures and progressive pulmonary metastases secondary to PC. Genetic analysis revealed a pathogenic variant in the CDC73 gene, indicating a hereditary predisposition. Following surgical resection, the patient experienced early biochemical relapse. Initial management with bisphosphonates, denosumab, and cinacalcet achieved temporary control of hypercalcemia. Upon radiological progression, lenvatinib therapy was initiated, resulting in 9 months of biochemical control and stabilization of both local disease and pulmonary metastases. However, discontinuation of denosumab and cinacalcet due to limited access led to a relapse of severe hypercalcemia and disease progression, necessitating the cessation of lenvatinib and transition to palliative care. This case underscores the diagnostic and therapeutic challenges of PC, highlights the potential role of targeted therapies like lenvatinib in advanced disease, and emphasizes the critical importance of sustained access to essential treatments.
Plain Language Summary
Parathyroid carcinoma (PC) is a rare cancer affecting the parathyroid glands, which control calcium levels in the blood. This type of cancer often leads to very high calcium levels, causing serious health problems like kidney disease and fragile bones. Treatment usually involves surgery, but the disease frequently comes back or spreads to other organs. We report the case of a 34-year-old man who had advanced PC with spread (metastases) to his lungs and bones. Despite surgery and standard treatments like zoledronic acid, denosumab, and cinacalcet to control his calcium levels, his disease continued to progress. We decided to use lenvatinib, a medication that targets specific pathways involved in tumor growth. Initially, lenvatinib helped control the disease for 9 months, stabilizing the size of his lung tumors and keeping calcium levels normal. However, after discontinuing other critical medications (denosumab and cinacalcet) due to limited availability, his disease rapidly progressed, and the calcium levels increased severely again. This case underscores a critical yet under-reported gap: while lenvatinib can temporarily restrain tumor growth, sustained disease control hinges on unbroken access to the full complement of calcium-lowering agents. When cinacalcet and denosumab were abruptly withdrawn for economic reasons, our patient experienced a swift recurrence of life-threatening hypercalcemia that ultimately rendered lenvatinib insufficient. These observations illustrate that the success of targeted therapy in advanced parathyroid carcinoma depends not only on the drug’s intrinsic antitumor activity but also on health-system factors that guarantee continuous, comprehensive treatment.
Introduction
PC was first described by De Quervain in 1904, and since then, over 1000 cases have been reported worldwide. 1 PC most commonly occurs as sporadic disease, although a minority of cases are inherited or associated with syndromes such as multiple endocrine neoplasia types 1 and 2A (MEN1, MEN2A), hyperparathyroidism-jaw tumor syndrome (HPT-JT) and isolated familial hyperparathyroidism.2,3 Pathogenic variants in CDC73 gene are identified in 47% to 60% of PC cases. CDC73, located on chromosome 1q31, encodes parafibromin, a tumor suppressor protein involved in regulating gene expression and inhibiting cell proliferation. Loss of parafibromin function, caused by pathogenic variants in CDC73, predisposes parathyroid tissue to malignant transformation. 4 Furthermore, CDC73 pathogenic variants have been associated with an increased risk of local recurrence and distant metastasis. 5 In the present case, a pathogenic variant in the CDC73 gene was identified.
Parathyroid tumors are rare endocrine neoplasms, occurring in 0.1% to 0.3% of the general population. Fewer than 1% are malignant, 6 and they account for <1% of cases of primary hyperparathyroidism (PHPT). 7 In the United States, the incidence of parathyroid carcinoma (PC) has increased to 11 cases/10 million people/year since 2001. 8 This increase rise may be attributed to more widespread screening for serum calcium levels, improved diagnostic methods, and increased life expectancy, although a true increase in incidence cannot be ruled out. 7
PC is most commonly diagnosed between the fifth and sixth decades of life.6,7,9 Hypercalcemia secondary to PC is associated with poor prognosis, is often refractory to treatment, and can lead to death, with estimated 5-year overall survival rates ranging from 78% to 85%. 10
Surgical resection is the first-line treatment for PC; however, the risk of persistent or recurrent disease is high, with a reported median disease-free survival of 8 to 10 months. 9 In this context, it is imperative to explore additional therapeutic options aimed at reducing tumor burden and controlling hypercalcemia and related symptoms. Pharmacological therapies—such as antiresorptives, cinacalcet, and systemic treatments—have emerged as alternatives, though available data are mostly limited to small series and case reports.
We present the case of a patient with persistent disease following parathyroidectomy and radiologic evidence of progressive pulmonary metastases. Treatment with Lenvatinib achieved 9 months of biochemical control and 8 months of structural stability. Initial biochemical control was attained with zoledronic acid, followed by monthly denosumab and cinacalcet. However, following the discontinuation of denosumab, the patient was readmitted to the emergency department due to severe hypercalcemia.
Case Report
A 34-year-old man with a history of chronic kidney disease and smoking and a family history of PHPT—his sister was diagnosed at 45 years of age; presented to the emergency department of Hospital San Juan de Dios in Rionegro, Colombia, with a 2-week history of low back pain following a fall. Initial laboratory tests revealed anemia, creatinine 3.55 mg/dl (KDIGO AKI stage II); estimated eGFR ≈22 ml/min/1.73 m2, and markedly elevated parathyroid hormone (PTH) levels at 1247 pg/ml. Serum calcium was initially within the normal range. Still, after initiating vitamin D replacement for a 25(OH)2D3 level of 13.47 ng/ml, hypercalcemia was unmasked, requiring management with intravenous fluid therapy (Figure 1 and Table 1).
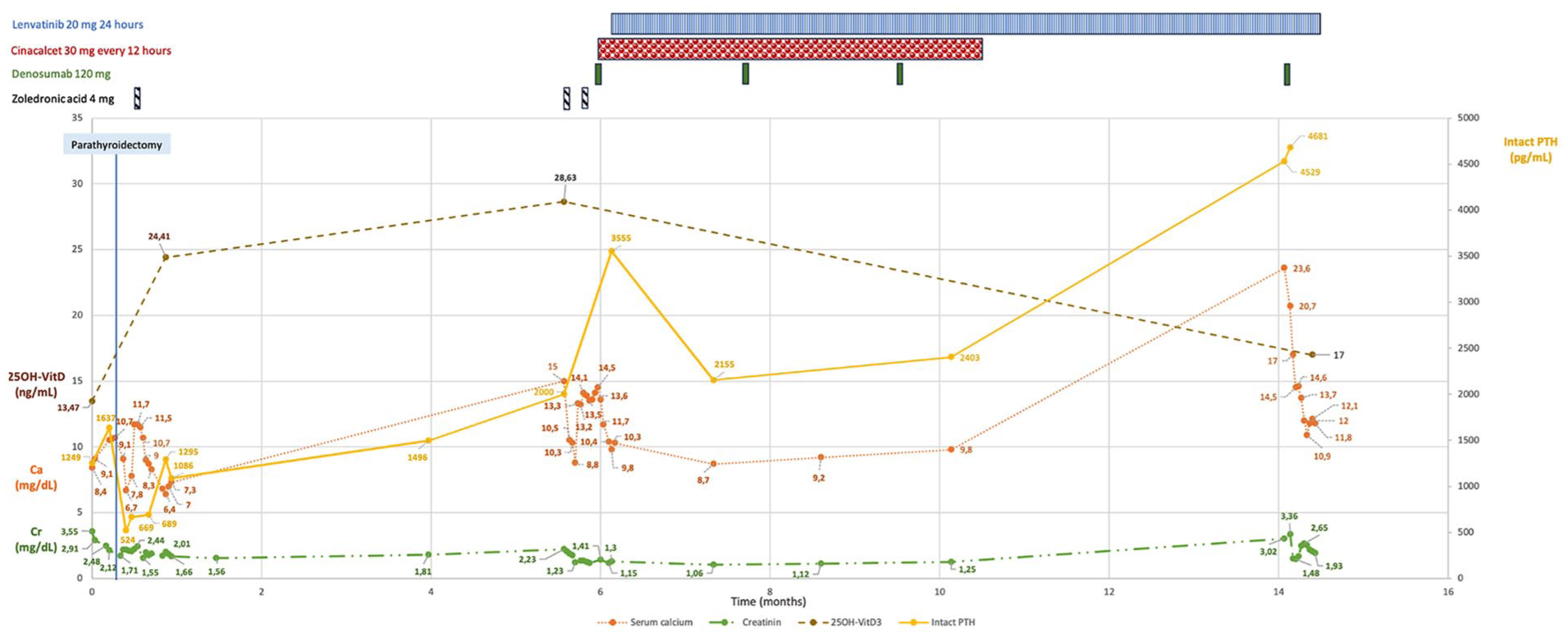
Summary of the clinical course and biochemical profile since the diagnosis of PC. Upon admission, laboratory tests revealed markedly elevated PTH levels and normal serum calcium, which progressively increased following correction of vitamin D deficiency. Hypercalcemia normalized after parathyroidectomy but recurred shortly thereafter, responding initially to intravenous zoledronic acid. The patient presented with acute kidney injury, with progressive improvement in creatinine levels following fluid therapy. Six months after diagnosis, a new episode of severe hypercalcemia occurred, unresponsive to additional doses of zoledronic acid. Normocalcemia was subsequently achieved with denosumab and cinacalcet. Due to rising PTH levels and radiological evidence of pulmonary metastases, treatment with lenvatinib was initiated. Continued use of denosumab and cinacalcet maintained stable calcium levels; however, following discontinuation of both agents, the patient was readmitted with another episode of severe hypercalcemia, which again responded to denosumab and intravenous fluids.
Initial Laboratory Tests.

Abbreviations: BUN, blood urea nitrogen; MCV, mean corpuscular volume; PTH, parathyroid hormone; TSH, thyroid-stimulating hormone; WBC, white blood cell; 25OH-VitD3, 25-hydroxy vitamin D3.
Computed tomography (CT) revealed lesions in the lumbosacral spine, multifragmented and displaced subcapital fractures, and multiple pulmonary nodules, predominating in lower lobes, the largest measuring 8 mm in the left lower lobe. HIV and tuberculosis were ruled out.
As part of the PHPT workup, technetium-99m-sestamibi (MIBI) scintigraphy showed no areas of radiotracer retention. Thyroid ultrasound identified a 26 × 25 × 36 mm TI-RADS 5 mass in the left lobe. Fine-needle aspiration biopsy of the nodule was classified as Bethesda II. Despite the findings, due to the suspicion of intrathyroidal parathyroid adenoma, the patient underwent parathyroidectomy. Intraoperatively, a large tumor mass with macroscopic invasion of surrounding tissues was discovered, prompting the decision to perform a left hemithyroidectomy with central neck dissection. Histopathology revealed neoplastic infiltration adjacent to the thyroid tissue, arranged in a nodular pattern, composed of neuroendocrine-like cells with frequent mitoses, necrosis (>70%), and vascular invasion. Immunohistochemistry was positive for GATA 3, synaptophysin, chromogranin, cytokeratin, and PTH; negative for thyroglobulin, calcitonin and TTF1, and showed a Ki-67 index of 20%. These findings were consistent with PC with vascular invasion. Parafibromin staining was unavailable at the institution. Next-generation sequencing revealed a heterozygous deletion del(1)(q31.2-1q31.3) (genomic coordinates chr1: 191399788-195082966), a pathogenic variant involving the CDC73 gene (cell division cycle).
Postoperatively, the patient’s PTH levels decreased to 524 pg/ml and serum calcium levels initially improved. However, 10 days later serum calcium rose to 12.4 mg/dl. A 4 mg intravenous dose of zoledronic acid normalized serum calcium, and the patient was discharged after 1 month of hospitalization (Figure 1).
Four months later, the patient was readmitted with severe hypercalcemia. Fluid therapy and 2 additional doses of zoledronic acid failed to correct the calcium levels. Management was escalated with a first dose of subcutaneous denosumab (120 mg) and cinacalcet (30 mg every 12 hours). Normocalcemia and symptom control were achieved at discharge, although PTH levels reached a peak of 2315 pg/ml. During hospitalization, CT and 18F-fluorocholine positron emission/computed tomography (PET/CT) revealed progressive pulmonary metastases from PC. As a result, treatment with oral lenvatinib (24 mg daily) was initiated, along with 2 additional monthly doses of denosumab. At a 3-month follow-up, the patient had normal serum calcium, decreased PTH levels, improved renal function, and stable lung metastases as confirmed by 18F-fluorocholine PET/CT.
The patient continued treatment with Lenvatinib. No further doses of zoledronic acid were administered. Cinacalcet and denosumab were suspended owing to patient affordability constraints in the Colombian health-care system. At 16 months after diagnosis, and 9 months into Lenvatinib therapy, the patient was readmitted to the emergency department with severe hypercalcemia and acute kidney injury. CT and 18F-fluorocholine PET/CT imaging revealed local disease progression and worsening pulmonary metastases. Based on these findings, a multidisciplinary team recommended discontinuing lenvatinib and continued palliative care, including analgesia, cinacalcet, and monthly denosumab (Figure 2).

18F-fluorocholine PET/CT images obtained before initiation of lenvatinib at 6 months after PC diagnosis (A–C), after 3 months of treatment (D–F), and after 9 months of therapy (G–I). At baseline, imaging revealed a lesion in the surgical bed of the left hemithyroidectomy, measuring 25 mm in maximum diameter with a peak SUV of 4.5 (A); pulmonary nodules with a maximum diameter of 11 mm and a peak SUV of 1.1 (B); and multiple, innumerable lytic bone lesions affecting both the axial and appendicular skeleton, with a maximum SUV of 6.0 (C). After 3 months of lenvatinib therapy, no significant changes were observed in the neck lesion (D), pulmonary nodules (E), or bone lesions (F), suggesting stable disease. At 9 months, imaging demonstrated disease progression with a doubling in volume of the neck lesion (G), increased size and number of pulmonary metastases, the largest measuring 16 mm (H), and the appearance of new lytic lesions in the calvarium, axial, and appendicular skeleton, along with increased metabolic activity in previously identified sites (I).
Discussion
The most prevalent clinical manifestations of PC include bone and renal complications, fatigue, a palpable cervical mass, and neuropsychiatric symptoms.1,7 The patient presented with multiple pathological fractures, low bone mineral mass, fibrocystic osteitis, long-standing mood and behavioral disturbances, and renal failure, all of which progressively improved with appropriate management and control of hypercalcemia.
Initial clinical evaluation typically includes biochemical and imaging studies. 15 PC is characterized by markedly elevated serum calcium and PTH levels. 7 Imaging studies assist in localization; definitive benign versus malignant distinction relies on histopathology, unless overt metastases are present. 2 Ultrasound shows 71% sensitivity and 100% specificity for detecting parathyroid lesions ⩾10 mm when correlated with surgical findings. 11 Additional modalities such as 4D-CT, scintigraphy, MRI, and PET/CT can be employed. 12 In our patient, despite a PTH level 10 times the upper limit of normal, MIBI scintigraphy failed to localize the lesion. False-negative MIBI can occur in poorly differentiated, cystic, or oxyphil-poor tumors; all 3 features have been linked to reduced radiotracer uptake. Ultrasound identified a mass in the left thyroid lobe, raising suspicion of MEN2A. For this reason, a biopsy of the thyroid mass was performed. However, immunohistochemistry of the parathyroidectomy specimen, along with negative plasma calcitonin and metanephrine levels, ruled out this diagnosis. It is important to note that fine-needle aspiration biopsy is generally not recommended in the evaluation of PC due to the risk of disrupting the tumor capsule and promoting tumor seeding. 13
The definitive diagnosis of PC is based on histopathological features, as defined in the 2022 WHO classification. 14 Vascular invasion, observed in this patient, is a key diagnostic criterion and a predictor of poor prognosis. It is associated with a 4-fold increased risk of death or recurrence at 5 years, and a 2.8- and 2.6-fold increased risk of overall recurrence and mortality, respectively. 13 Among immunohistochemistry markers, parafibromin loss, galectin-3 positivity, and Ki-67 >5%, have the strongest diagnostic utility for PC. 14 In our patient, Ki-67 was 20%, parafibromin testing was not available, but the presence of a CDC73 pathogenic variant is highly predictive of parafibromin-deficient parathyroid neoplasms. These mutations characteristically occur in parathyroid carcinoma associated with HPT-JT syndrome or familial forms of primary hyperparathyroidism 14 ; the positive family history in our patient supports the latter scenario. Aside from the CDC73 deletion, the targeted 50-gene panel identified no additional actionable alterations—including MEN1, RET, or CDKN1B.
Disease trajectory is highly variable: median overall survival up to 14.3 years has been reported, yet aggressive cases such as ours progress rapidly. Surgery is the cornerstone of initial treatment and is also indicated for local recurrence that occurs even in the 40% to 60% and metastatic disease. 7 However, PC carries a recurrence rate of 40% to 60% and a median overall survival of 14.3 years. 15 These outcomes may be influenced by a preoperative misdiagnosis rate exceeding 80%, which often prevents the performance of radical surgery—an approach that has been shown to reduce recurrence, decrease the need for reoperation, and improve survival. 10 Distant metastases are presented at diagnosis in 10% to 30% of patients, most commonly affecting lungs, bones, and liver. 16
There is limited evidence regarding the effectiveness and timing of radiotherapy, chemotherapy, or immunotherapy. 7 The major cause of morbidity in PC is refractory hypercalcemia due to inoperable recurrent or metastatic lesions. Standard measures include intravenous fluids, diuretics, and bisphosphonates. When these are insufficient, calcimimetic agents such as cinacalcet are recommended.7,15 The use of denosumab for hypercalcemia management is supported by case reports. 17
Recently, tumor-directed systemic therapies have emerged as a potential option for PC. 18 The use of sorafenib is based on the overexpression of vascular endothelial growth factor receptors (VEGFR) and platelet-derived growth factor receptors (PDGFR) in PC, both of which are involved in tumor growth, invasion, and metastasis. Approximately 50% of PC patients respond to sorafenib. 19 More recently, Lenvatinib has shown promising results in PC cases. Teleanu et al 18 reported a case of a 51-year-old man with PC who, 4 years after parathyroidectomy, developed pulmonary and skull base metastases. Treatment with lenvatinib achieved biochemical control during 2 separate treatment periods. Similarly, Ito et al 20 described a 61-year-old woman with pulmonary and spinal metastases and VEGFR2-overexpression. Lenvatinib achieved rapid biochemical control and reduced metastases but had to be discontinued after 2 months due to hematuria and thrombocytopenia. He later developed local recurrence, and died. Finally, immunotherapy with pembrolizumab or nivolumab can be considered as an alternative management in patients with PC refractory to other lines of treatment.21,22
In our case, the patient experienced an initial rapid response to lenvatinib, with 9 months of stable structural disease. However, progression of both local and metastatic disease was later documented. Our experience, in line with that of other authors, highlights the importance of combining cinacalcet with bisphosphonates or denosumab—often at increasingly frequent dosing intervals—to effectively manage hypercalcemia in advanced PC.
Conclusion
To our knowledge, the case we report is the first of lenvatinib management in PC in Latin America. After 3 months of therapy, the drug has been well tolerated.
Afforded initial control but hypercalcemia and structural progression recurred after 9 months, and the patient continued palliative care.
Footnotes
Acknowledgements
Our thanks to the Endocrinology Service of the Hospital San Juan de Dios, Rionegro, Colombia for their support in the patient’s diagnostic analysis.
Authors’ Note
Wilfredo A. Rivera is now affiliated to Endocrinology Department, IPS Neurum, Medellín, Colombia.
Ethical Considerations
This study was approved by the ethics committee of Hospital San Juan de Dios, Rionegro, Colombia, and was conducted in accordance with the Declaration of Helsinki.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Author Contributions
W.A.R. conceptualization, data curation, writing—original draft, writing—review and editing. M.J.R. conceptualization, data curation, writing—original draft. A.R. conceptualization, supervision, writing—review and editing.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors declare that they have no conflicts of interest for the writing and publication of this article. A.R. has received fees as a speaker or advisory board member from the Colombian Association of Endocrinology, Diabetes, and Metabolism, ROCHE, Knightx, Bayer, Novo Nordisk, Biotoscana, PTC, Amgen, Recordati, Amryt, Valentech, Ipsen, Pfizer, Ultragenyx, Sanofi, Chiesi, Biosidius, Lilly.
Data Availability Statement
All data generated during this study are included in this published article.