
Abstract
Background:
Transcription factor 7-like 2 (TCF7L2) variants seem to affect diabetes susceptibility through β-cell dysfunction, underlying basis of which has been considered to be β-cell dedifferentiation rather than apoptotic β-cell death. The Extracellular regulated protein kinases/Mitogen-activated protein kinase signaling pathway (ERK/MAPK signaling pathway) has been confirmed to be significantly associated with multiple cellular process, including cellular dedifferentiation. However, the effects of TCF7L2 on β-cell function and ERK/MAPK signaling pathway are poorly understood.
Objectives:
This study aimed to elucidate the regulation of TCF7L2 in β-cell function and ERK/MAPK signaling pathway, which further participate in glucose metabolism and diabetes progression.
Methods:
After transfection of TCF7L2 siRNA and lenti-TCF7L2 plasmids, the activation of ERK/MAPK signaling and β-cell dedifferentiation were evaluated respectively. Six week-old male db/db mice were randomly grouped and fed a normal or high-fat diet, and then pancreatic level of TCF7L2 protein were measured respectively when the mice were fed to 8, 12, and 16 weeks of age. Furthermore, the contributions of TCF7L2 to ERK/MAPK signaling and glucose metabolism were investigated in a β-cell-specific TCF7L2 deletion mice model (TCF7L2β−/−).
Results:
The results demonstrated that impaired TCF7L2 induces β-cell dedifferentiation and decreases insulin secretion of MIN6 cells via ERK/MAPK signaling pathway. Consistently, decreased pancreatic TCF7L2 protein in parallel with reduced functional β-cells were observed in db/db mice after weeks of normal or high-fat diet. However, the differences between were only significant when the mice were fed to 12 weeks of age. After weeks of high-fat diet feeding, impaired glucose tolerance and increased activation of ERK/MAPK signaling were simultaneously observed in TCF7L2β−/− mice.
Conclusion:
The study indicate that the induction of β-cell dedifferentiation mediated by ERK/MAPK signaling pathway might be an essential component of TCF7L2 variants in the development of diabetes.
Plain Language Summary
Transcription factor 7-like 2 (TCF7L2) variants seem to affect diabetes susceptibility through β-cell dysfunction, underlying basis of which has been considered to be β-cell dedifferentiation rather than apoptotic β-cell death. Nowadays, how TCF7L2 participates in β-cell dedifferentiation and how pancreatic TCF7L2 protein changes during diabetes progression are poorly understood. The Extracellular regulated protein kinases/Mitogen-activated protein kinase signaling pathway (ERK/MAPK signaling pathway) has been confirmed to be significantly associated with multiple cellular process, including cellular dedifferentiation. Here, we demonstrated that impaired TCF7L2 induces β-cell dedifferentiation and decreases insulin secretion of MIN6 cells via ERK/MAPK signaling pathway. Consistently, declined pancreatic TCF7L2 protein in parallel with reduced mature β-cells were observed in db/db mice after weeks of normal or high-fat diet feeding. Furthermore, the role of TCF7L2 in dedifferentiation and glucose homeostasis were validated in mice with β cell-specific TCF7L2 deletion (TCF7L2β−/−). In conclusion, the study indicate that the induction of β-cell dedifferentiation mediated by ERK/MAPK signaling pathway might be an essential component of TCF7L2 variants in the development of diabetes.
Introduction
Type 2 diabetes mellitus (T2DM) is one of the most common metabolic diseases and is characterized by chronic hyperglycemia mainly due to pancreatic β-cell destruction or dysfunction. 1 The mechanism underlying β-cell failure in diabetes is controversial and is almost exclusively attributed to apoptotic β-cell death. However, an important concept regarding β-cell failure has been proposed, which is not only due to apoptotic β-cell death but also the differentiation of insulin-producing mature β-cells into endocrine progenitor cells. 2 This process, known as dedifferentiation, leads to severe dysfunction of insulin secretion, which is a major contributor to hyperglycemia in diabetes.3,4 There are reasons to think that dedifferentiation might be reversible because of the prevention or reversion of β-cell dysfunction by good glycemic control in T2DM patients.5,6 In addition, insulin-producing mature β-cells may be converted to other endocrine cells, including α-cells.
Various transcription factors are involved in pancreatic β-cell differentiation and the maintenance of mature β-cell function. During the dedifferentiation process, β-cells lose their identity and function with a decrease in the expression of β-cell-specific markers such as MafA, Ucn3, FoxO1, and Glp1r.7,8 These dedifferentiated β-cells express endocrine progenitor cell markers such as Neurog3, Sox9, Hes1, and L-Myc. 9
TCF7L2 (transcription factor 7-like 2), a basic component of the Wnt signaling pathway, is a genetic variant that is considered to be definitively associated with T2DM. This association has consistently been replicated in multiple populations with diverse genetic origins.10 -12 However, the mechanism by which abnormal TCF7L2 activity contributes to β-cell dysfunction remains unclear. Previous studies have demonstrated that TCF7L2 modulates β-cell function by affecting β-cell growth or differentiation.13,14 Evidence also suggests that TCF7L2 may mediate β-cell proliferation, and silencing of TCF7L2 in cell lines and primary islets leads to increased apoptosis.15,16 As β-cell dedifferentiation is also considered a major contributor to β-cell dysfunction, we investigated the effect of TCF7L2 on β-cell dedifferentiation and whether impaired insulin secretion in diabetes can be explained by an altered β-cell differentiation state.
The Extracellular regulated protein kinases/Mitogen-activated protein kinase signaling pathway (ERK/MAPK signaling pathway), a most thoroughly studied MAPK signaling cascade, has been confirmed to be significantly associated with multiple cellular process and signal transduction network.17,18 Previous studies have demonstrated that the ERK/MAPK signaling pathway plays an important role in cellular dedifferentiation.19,20 Interestingly, previous experiments also found that impaired TCF7L2 significantly reduced ERK phosphorylation in human colorectal cancer cells, and treatment with the ERK phosphorylation inhibitor U0126 decreased TCF7L2-induced endogenous LCN2 expression in ESCC cells.21,22 These findings raise the possibility that TCF7L2 may serve as a modulator of ERK/MAPK signaling.
In this study, we investigated the specific role of TCF7L2 in β-cell dedifferentiation and glucose metabolism in the MIN6 and db/db mice. Combined with the model of TCF7L2β−/− mice, we confirmed that the effects of TCF7L2 on β-cell function and glucose homeostasis were mediated by the activation of ERK/MAPK signaling pathway.
Methods
Cell lines and antibodies
MIN6, a mouse pancreatic β-cell line, was generously provided by Dr. Yang of Joslin Diabetes Center (Harvard Medical School, Boston, MA, USA). All cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Cells were synchronized using serum-free medium when they reached 65% attachment.
Antibodies targeting the following proteins were obtained from Cell Signaling Technology Inc. (Danvers, MA, USA): Hes1 (Cat. no. 11988) and FoxO1 (cat. no.2880), and total ERK (cat. no.4696) and phospho-ERK (cat. no. 4370). The TCF7L2 antibody (cat. no. H00006934-M06) was purchased from Novus Biologicals Inc. A specific inhibitor of ERK phosphorylation (U0126) was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Cell transfection
When the cells were 60% to 70% confluent, they were transfected with the lenti-TCF7L2 plasmid or siRNA targeting TCF7L2. Cell transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocol.
Animals
Male db/db mice (6-week old; background strain Cg-Dock7m+/+ Leprdb, Charles River, China) was housed in a light (12 hours/12 hours on/off) and temperature-controlled environment. The mice were fed a normal diet (ND; Harlan Teklad Rodent Diet 8604, containing 12.2, 57.6%, and 30.2% energy from fat, carbohydrate, and protein, respectively) or high-fat diet (HFD; Surwit Research Diets, New Brunswick, NJ, USA, containing 58, 26%, and 16% energy from fat, carbohydrate, and protein, respectively). All the animal protocols were approved by the Animal Welfare and Ethics Committee at Fudan University in agreement to NIH animal care guidelines.
Intraperitoneal glucose tolerance test
For the intraperitoneal glucose tolerance test (IPGTT), mice were fasted 12 hours overnight and injected intraperitoneally with glucose at a dose of 2 g/kg body weight. Blood samples were obtained at 0, 30, 60, 90, and 120 minutes. The glucose concentration was determined using a glucometer (Roche, Indianapolis, IN, USA).
Western blots
Extracted protein was lysated and measured by using a BCA Protein Assay Kit (Beyotime Inc., China). Protein samples were separated and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked in 4% milk for 2 hours at room temperature and then incubated overnight at 4°C with primary antibodies. 23 Band density were analyzed by using LabWork 4.5 image software.
Real-time quantitative PCR
Total RNA was extracted from the cell lines using TRIzol Reagent (Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions. Reverse transcription was performed using a Reverse Transcription Kit (Thermo Fisher Scientific, Inc.). qPCR was performed in triplicate using the SYBR Green Master Mix (Takara Bio, Inc., Otsu, Japan) on an ABI 7000 Sequence Detection System (Thermo Fisher Scientific, Inc.). All the primers used are listed in Table 1.
Primers used for target amplification in real-time PCR.

Immunohistochemistry
Paraformaldehyde-fixed pancreatic tissues (4%) were dehydrated, paraffin-embedded, sectioned sequentially, dewaxed, and hydrated for antigen repair. Following overnight incubation with anti-TCF7L2 (dilution, 3 µg/ml) at 4°C, the sections were rinsed in PBS and incubated with the secondary antibody at room temperature for 30 minutes. After blocking, the sections were observed under a microscope.
Statistical analysis
Data are presented as the mean ± standard deviation and were analyzed using Graph Pad software (version 6.0c; Graph Pad Software, Inc., La Jolla, CA, USA). P < .05 was considered statistically significant.
Result
Impaired TCF7L2 induced β-cells dedifferentiation and decreased insulin secretion of MIN6 cells
Mature β-cell marker (FoxO1) and endocrine progenitor marker (Hes1) levels were measured after TCF7L2 siRNA plasmid transfection. The increased expression of Hes1 and decreased expression of FoxO1 indicated that impaired TCF7L2 induced β-cell dedifferentiation (Figure 1A-C), and destroyed β-cell function further resulted in decreased insulin secretion of MIN6 cells (Figure 1D).

Impaired TCF7L2 induced β-cells dedifferentiation and decreased insulin secretion of MIN6 cells. (A) The expressions of TCF7L2, Hes1 and FoxO1 were measured respectively by western blot after the transfection of TCF7L2 siRNA plasmid. (B) The expression of Hes1 was measured by real-time PCR after the transfection of TCF7L2 siRNA plasmid. (C) The expression of FoxO1 was measured by real-time PCR after the transfection of TCF7L2 siRNA plasmid. (D) The insulin concentration was evaluated by ELISA after the transfection of TCF7L2 siRNA plasmid. (E) The expressions of TCF7L2, Hes1 and FoxO1 were measured respectively by western blot after the transfection of lenti-TCF7L2 plasmid. (F) The expression of Hes1 was measured by real-time PCR after the transfection of lenti-TCF7L2 plasmid. (G) The expression of FoxO1 was measured by real-time PCR after the transfection of lenti-TCF7L2 plasmid. (H) The insulin concentration was evaluated by ELISA after the transfection of lenti-TCF7L2 plasmid. (n = 3 samples per groug, **P < .01 and *P < .05 as indicated).
Following the transfection of lenti-TCF7L2 plasmid, changes of β-cell dedifferentiation and insulin secretion were completely reversed (Figure 1E-H). GAPDH was used as an internal control for normalization. The primers used for target amplification are listed in Table 1.
The effects of TCF7L2 on β-cell dedifferentiation and insulin secretion were mediated by ERK/MAPK signaling pathway
To determine the effects of TCF7L2 on ERK/MAPK signaling, total and phosphorylated ERK were measured after transfection with TCF7L2 siRNA plasmid. The data suggested that impaired TCF7L2 led to increased ERK phosphorylation, which indicating increased activation of ERK/MAPK signaling pathway (Figure 2A). However, changes in Hes1, FoxO1, and insulin secretion induced by impaired TCF7L2 were completely attenuated when ERK phosphorylation was blocked by U0126 (Figure 2B-D), indicating that the effects of TCF7L2 on β-cell dedifferentiation and insulin secretion were mediated by the ERK/MAPK signaling pathway.
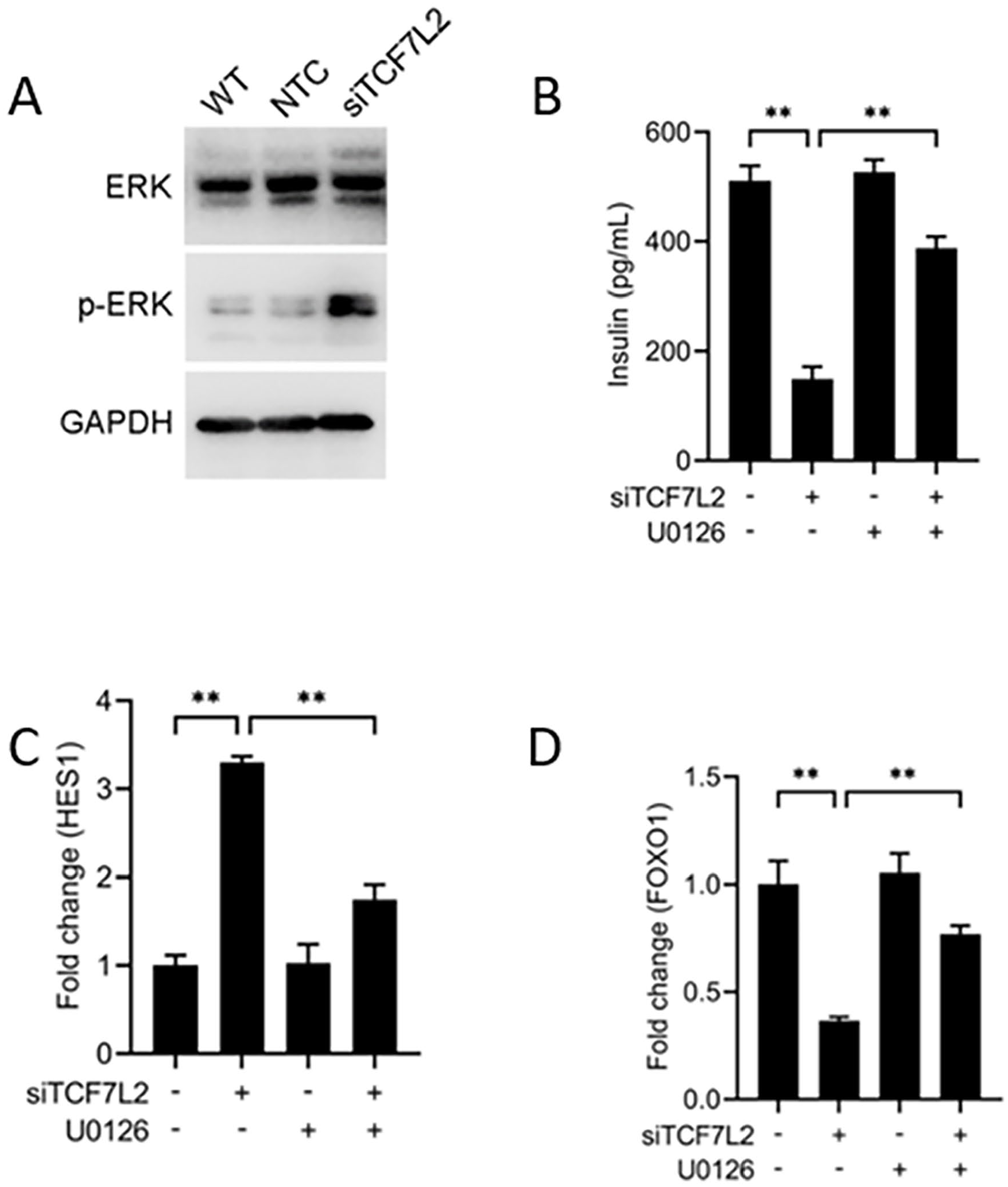
The effects of TCF7L2 on β-cell dedifferentiation and insulin secretion were mediated by ERK/MAPK signaling pathway. (A) The expressions of total and phosphorylated ERK were evaluated respectively by western blot after TCF7L2 siRNA plasmid transfection. (B) The insulin concentrations in supernatant were measured by ELISA when MIN6 cells were cultured with 10 µM ERK inhibitor U0126 for 30 minutes prior to TCF7L2 siRNA plasmid transfection. (C) The expressions of Hes1 were evaluated by real-time PCR when MIN6 cells were cultured with 10 µM ERK inhibitor U0126 for 30 minutes prior to TCF7L2 siRNA plasmid transfection. (D) The expressions of Foxo1 were evaluated by real-time PCR when MIN6 cells were cultured with 10 µM ERK inhibitor U0126 for 30 minutes prior to TCF7L2 siRNA plasmid transfection. (n = 3 samples per groug, **P < .01 as indicated).
Reciprocal changes of pancreatic TCF7L2 protein during diabetes progression
Six week-old male db/db mice were randomly grouped and fed a normal or high-fat diet. After weeks of feeding, body weight and fasting glucose levels were measured, both of which increased over time during the procedure. However, HFD-fed mice showed higher body weight and glucose levels than those fed the normal diet (Figure 3A and B). The differences were significant when the mice were fed to 12 weeks of age.

Reciprocal changes of pancreatic TCF7L2 protein during diabetes progression. (A) The body weight of db/db mice (n = 5) was measured after weeks of normal or high-fat diet feeding. (B) Fasting glucose levels in db/db mice (n = 5) were measured after weeks of a normal or high-fat diet. (C) Changes in pancreatic TCF7L2 and FoxO1 levels in db/db mice were measured using western blotting after weeks of normal or high-fat diet feeding. (D) Quantification of TCF7L2 expression in db/db mice after weeks of a normal or high-fat diet. (E) Quantification of FoxO1 expression in db/db mice after weeks of a normal or high-fat diet. (F) Local expression of TCF7L2 in islets was measured by immunohistochemistry after weeks of high-fat diet feeding. (n = 3 samples per groug, **P < .01 as indicated).
To determine how TCF7L2 levels change during diabetes, pancreatic TCF7L2 levels were measured by western blotting after weeks of feeding, which decreased with time in both normal and high-fat diet groups. However, the expression of FoxO1, whose localization was restricted to β-cells, also declined after weeks of feeding. (Figure 3C). Interestingly, the expression of TCF7L2 and FoxO1 was significantly higher in mice fed with high-fat diet, but the difference was only significant at 12 weeks. (Figure 3D and E).
Pancreatic tissue of mice after weeks of high-fat diet feeding was obtained and sliced to investigate the local expression of TCF7L2 in the islets using immunohistochemistry. The data indicated that a high-fat diet could significantly diminish islet expression of TCF7L2, which was barely reduced when the mice were fed to 16 weeks of age. (Figure 3F).
TCF7L2 deficiency destroyed glucose tolerance and insulin secretion to some extent
To examine the β-cell specific function of TCF7L2 in diabetes, we mated TCF7L2fl/fl and mip-cre ERT2 mice to generate mice with a β-cell specific TCF7L2 deletion (referred to as TCF7L2β−/−). Genotypes of the candidate mice were confirmed by PCR using the genome obtained from the tail. Shown in Figure 4A, the wild type present one band with 476 bp and homozygote type present one band with 586 bp by using TCF7L2 primer, so 2 bands with 476 and 586 bp represent heterozygote type. As indicated, both TCF7L2fl/fl and TCF7L2β−/− mice should present one band with 586 bp in PCR by using TCF7L2 primer, and TCF7L2β−/− mice should also present a band with 200 bp by using Mip-cre primer, but not TCF7L2fl/fl mice. Here, H20 served as negative control (Figure 4B).

Weekly body weight of TCF7L2fl/fl and TCF7L2β−/− mice were recorded. (A) Genotype of the candidate mice were confirmed by PCR using TCF7L2 primer. (Wild type: one band with 476 bp; Heterozygote: two bands with 476 and 586 bp; Homozygote: one band with 586 bp.) WT: Wild type, H20: Negative control. (B) Genotype of the candidate mice were confirmed by PCR using Mip-cre primer. (Target: 300 bp; Control: 200 bp.) WT: Wild type, H20: Negative control. (C) Body weight of TCF7L2fl/fl (n = 6) and TCF7L2β−/− (n = 6) mice were measured weekly from 8 to 12 weeks of age, after normal diet feeding. (D) Body weight of TCF7L2fl/fl (n = 6) and TCF7L2β−/− (n = 6) mic were measured weekly from 8 to 12 weeks of age, after high-fat diet feeding. All data are represented as mean ± SEM, and statistical analyses were performed using Student’s t-test (**P < .01, *P < .05).
Primers used for target amplification were: TCF7L2 CKO: 5’-ATCCACCTCCGCACTTAC-3’ (forward), 5’-TGGGCTCACAATAAACATCC-3’ (reverse); Mip-cre: 5’-CCTGGCGATCCCTGAACATGTCCT-3’ (Transgene Forward), 5’-TGGACTATAAAGCTGGTGGGCAT-3’ (reverse); Internal Positive Control: 5’-CAAATGTTGCTTGTCTGGTG-3’ (forward) and 5’-GTCAGTCGAGTGCACAGTTT-3’ (reverse).
Weekly body weights were recorded from 8 to 12 weeks of age. There was no difference in body weight between TCF7L2fl/fl and TCF7L2 β−/− mice fed on normal diet (Figure 4C). However, TCF7L2β−/− mice showed higher body weight than TCF7L2fl/fl mice after weeks of high-fat diet feeding, and the differences became significant after 11 weeks (Figure 4D).
To determine the role of TCF7L2 in glucose metabolism, we performed glucose tolerance tests in TCF7L2fl/fl and TCF7L2β−/− mice. The data demonstrated that TCF7L2 deletion in β-cells significantly disrupted glucose homeostasis when the mice were fed for 12 weeks, regardless of whether they were fed on normal or high-fat diet (Figure 5A-C). However, TCF7L2 deficiency only decreased insulin secretion in mice fed a high-fat diet for 12 weeks, whereas mice fed a normal diet showed no significant difference (Figure 5D-F).

TCF7L2 deficiency destroyed glucose tolerance and insulin secretion to some extent. (A) Glucose tolerance tests on TCF7L2fl/fl (n = 4) and TCF7L2β −/− (n = 5) mice at 8 weeks. (B) Glucose tolerance tests on TCF7L2fl/fl (n = 6) and TCF7L2β−/− (n = 6) mice at 12 weeks, after normal diet feeding. (C) Glucose tolerance tests on TCF7L2fl/fl (n = 6) and TCF7L2β−/− (n = 6) mice at 12 weeks, after high-fat diet feeding. (D) Insulin secretion of TCF7L2fl/fl (n = 4) and TCF7L2β−/− (n = 5) mice at 8 weeks. (E) Insulin secretion of TCF7L2fl/fl (n = 6) and TCF7L2β−/− (n = 6) mice at 12 weeks, after normal diet feeding. (F) Insulin secretion of TCF7L2fl/fl (n = 6) and TCF7L2β−/− (n = 6) mice at 12 weeks, after high-fat diet feeding. All data are represented as mean ± SEM, and statistical analyses were performed using Student’s t-test (**P < .01, *P < .05).
TCF7L2 deficiency induce β-cell dedifferentiation and activation of ERK/MPAK signaling
To investigate the effects of TCF7L2 deficiency on β-cell dedifferentiation, the expression of pancreatic Hes1 and FoxO1 in TCF7L2fl/fl and TCF7L2β−/− mice was measured by western blotting when the mice were fed a high-fat diet to 12 weeks of age. The increased expression of Hes1 and decreased expression of FoxO1 indicating increased dedifferentiation of β-cells in TCF7L2β−/− mice. However, we also observed increased ERK phosphorylation in TCF7L2β−/− mice, which confirmed our previous hypothesis (Figure 6).

TCF7L2 deficiency induce β-cell dedifferentiation and activation of ERK/MPAK signaling. (A) Genotypes of TCF7L2fl/fl and TCF7L2β−/− mice were confirmed by PCR. (B) Expressions of Hes1, FoxO1 and p-ERK in TCF7L2fl/fl and TCF7L2β−/− mice were measured respectively by western blotting when the mice were fed with high-fat diet to 12 weeks of age.
Discussion
Of the possible SNPs that have been associated with T2DM, TCF7L2 is the most potent locus for diabetes risk and the first locus to have been repeatedly demonstrated by genome-wide association studies. 24 Previous studies indicated TCF7L2 target genes are tissue and context-specific, it specifically binds to multiple genes that are important in glucose metabolism in hepatocytes and a list of validated TCF7L2 cardiac-specific target genes is also identified. 25 However, TCF7L2 variants have been shown to affect diabetes susceptibility through β-cell dysfunction, because TCF7L2 silencing exerts a strong inhibitory effect on glucose-mediated insulin secretion. 26 TCF7L2 itself promotes beta cell proliferation, protects against apoptosis, and improves insulin secretion. 27 As the underlying basis of β-cell dysfunction is considered to be β-cell dedifferentiation rather than apoptotic β-cell death, 5 we investigated whether TCF7L2 variants participate in regulating β-cell dedifferentiation, which further leads to impaired insulin secretion in diabetes.
It has been demonstrated that β-cell dedifferentiation may be linked to the decreased expression of specific mature β-cell markers and increased levels of endocrine progenitor cell markers. In the present study, we found that impaired TCF7L2 reduced the expression of FoxO1, whose localization is restricted to β-cells, and its decline in T2DM patients is paralleled by the loss of insulin immunoreactivity. 28 We also observed an increased expression of Hes1, an endocrine progenitor cell marker, after TCF7L2 siRNA transfection. Taken together, impaired TCF7L2 expression leads to β-cell dedifferentiation, which represents an alternative mechanism for explaining the loss of β-cell function in diabetes.
Previous data demonstrated that both SAPK/JNK and ERK/MAPK signaling play essential roles in diabetes, and increased ERK phosphorylation in macrophages mostly occurs by exerting a protective effect in a chronic high glucose environment.29,30 There is also evidence suggesting that sustained activation of ERK/MAPK signaling in adipocytes is associated with the pathogenesis T2DM and its selective blockade by MEK inhibitors may be a promising approach for the treatment of insulin resistance in diabetes. 31 However, little is known about the mechanism by which the ERK/MAPK signaling pathway contributes to β-cell dysfunction, and the effects of TCF7L2 on ERK/MAPK signaling are controversial. Previous studies have demonstrated that stable knockdown of TCF7L2 significantly reduced ERK phosphorylation in acute lymphoblastic leukemia, and RAS-induced activation of ERK signaling in colorectal cancer cells was mediated by TCF7L2-regulated gene expression.22,32 These data indicate that TCF7L2 may serve as a positive modulator of the ERK/MAPK signaling pathway. However, our experiment demonstrated that impaired TCF7L2 expression leads to sustained activation of ERK/MAPK signaling. These contradictory results can potentially be explained by the different genetic backgrounds of different cell lines. However, the effects of TCF7L2 on β-cell dedifferentiation and insulin secretion were completely attenuated following administration of the ERK phosphorylation inhibitor U0126, indicating that β-cell dedifferentiation induced by impaired TCF7L2 was mediated by ERK/MAPK signaling, which further led to a loss of functioning β-cells.
It is generally believed that developing therapies that modulate TCF7L2 expression may provide opportunities to treat T2DM patients carrying a TCF7L2 risk allele. Multiple previous studies have investigated the changes in TCF7L2 in diabetes, which were observed to be almost undetectable in pancreatic sections of T2DM patients, 33 and its protein expression was consistently impaired in various rodent models of T2DM.27,34 The role of TCF7L2 in β-cell dedifferentiation and changes in pancreatic TCF7L2 protein during diabetes progression is poorly understood. As animal models of high-fat diets are widely used in the study of metabolic diseases, we investigated the changes in TCF7L2 in db/db mice after weeks of normal or high-fat diet feeding. Consistent with previous findings, 33 declined TCF7L2 and FoxO1 protein levels were observed simultaneously during the procedure. However, higher expression levels of TCF7L2 and FoxO1 were observed in mice fed with a high-fat diet, but the difference was only significant when the mice were fed to 12 weeks of age, which might be explained by aggravated β-cell dysfunction in diabetes progression. Long-term glucose toxicity induced β-cell failure when the mice were fed to 16 weeks of age, no matter on normal or high-fat diet.
Previous studies have shown that TCF7L2 KO mice died shortly after birth, but β-cell-specific knockout did not affect their survival. 35 To investigate the β-cell-specific function of TCF7L2 in diabetes, we generated TCF7L2β−/− mice by mating TCF7L2fl/fl and mip-cre ERT2 mice. TCF7L2β−/− mice showed markedly impaired glucose homeostasis and insulin secretion after weeks of feeding, especially on a high-fat diet, which might be explained by aggravated β-cell dysfunction induced by TCF7L2 deletion. Since β-cell dedifferentiation is considered the underlying basis of β-cell dysfunction, our analyses demonstrated that TCF7L2β−/− mice underwent increased dedifferentiation after TCF7L2 deletion. In addition, the increased phosphorylation of ERK detected in TCF7L2β−/− mice further confirmed our hypothesis that β-cell dedifferentiation induced by impaired TCF7L2 is mediated by ERK/MAPK signaling activation.
The limitations of this study are as follows. Firstly, the contributions of TCF7L2 to β-cell dedifferentiation and insulin secretion have been confirmed to be mediated by ERK/MAPK signaling pathway in MIN6 cells. By using mice models of db/db and β-cell specific TCF7L2 deletion(TCF7L2β−/−), the contribution of TCF7L2 to glucose metabolism has been reconfirmed. However, although the changes of pancreatic Fox1 and Hes1, which serving as dedifferentiation marker, have been investigated in TCF7L2β−/− mice, visual labeling of dedifferentiated cells may be more convincing and further more morphological features need to be studied. Secondly, we only investigated the changes of glucose metabolism in mice models, but ignored most of the possible confounding factors such as lipid metabolism, which may affect the final observation. Furthermore, the power analysis for sample size calculation was not done, which may result in insufficient statistical power or uncertainties in effect size estimation.
Conclusion
The present study demonstrated that pancreatic β-cell dedifferentiation mediated by ERK/MAPK signaling may be an essential component of TCF7L2 variants in the development of diabetes. Considering that T2DM is a reversible disease, it is possible to improve β-cell function by re-establishing cellular maturation and glycemic control during disease progression. Further studies will address pancreatic β-cell dedifferentiation and its underlying mechanisms that may occur.