
Abstract
Background:
Type 2 diabetes mellitus (T2DM) is the most common metabolic disorder. The aim of the present investigation was to identify gene signature specific to T2DM.
Methods:
The next generation sequencing (NGS) dataset GSE81608 was retrieved from the gene expression omnibus (GEO) database and analyzed to identify the differentially expressed genes (DEGs) between T2DM and normal controls. Then, Gene Ontology (GO) and pathway enrichment analysis, protein-protein interaction (PPI) network, modules, miRNA (micro RNA)-hub gene regulatory network construction and TF (transcription factor)-hub gene regulatory network construction, and topological analysis were performed. Receiver operating characteristic curve (ROC) analysis was also performed to verify the prognostic value of hub genes.
Results:
A total of 927 DEGs (461 were up regulated and 466 down regulated genes) were identified in T2DM. GO and REACTOME results showed that DEGs mainly enriched in protein metabolic process, establishment of localization, metabolism of proteins, and metabolism. The top centrality hub genes APP, MYH9, TCTN2, USP7, SYNPO, GRB2, HSP90AB1, UBC, HSPA5, and SQSTM1 were screened out as the critical genes. ROC analysis provides prognostic value of hub genes.
Conclusion:
The potential crucial genes, especially APP, MYH9, TCTN2, USP7, SYNPO, GRB2, HSP90AB1, UBC, HSPA5, and SQSTM1, might be linked with risk of T2DM. Our study provided novel insights of T2DM into genetics, molecular pathogenesis, and novel therapeutic targets.
Keywords
Introduction
Type 2 diabetes mellitus (T2DM) is a complex metabolic disorder and is characterized primarily by a decrease in insulin secretion, typically accompanied by insulin resistance. 1 Globally, it is predicted that 25 million adults (20-79 years) have diabetes, projected to reach 629 million by 2045 and is the ninth leading cause of death.2,3 T2DM is mainly associated with macrovascular complications include stroke, coronary artery disease and peripheral arterial disease, and microvascular complications include diabetic retinopathy, diabetic nephropathy, and diabetic neuropathy, and non-vascular diabetes complications include nonalcoholic fatty liver disease, psychiatric disease, obesity, cancer, cognitive impairment, infections, and disability. 4 There are several important risk factors for T2DM, such as age, sex, family history of diabetes, hypertension, obesity, abdominal obesity, stress in the workplace or home, a sedentary lifestyle, smoking, insufficient fruit and vegetable consumption, physical activity, genetic, and environmental causes. 5 Our understanding of the occurrence and development mechanism of T2DM has been greatly improved; however, the cause and potential molecular mechanism of T2DM are still unclear. 6 Therefore, it is necessary to identify key genes and pathways for understanding the molecular mechanism and discovering potential biomarkers for T2DM.
In recent decades, more and more researchers have devoted themselves to exploring the potential mechanisms for progression of T2DM. Recent investigation have shown that key biomarkers, such as HHEX, CDKN2A/B, and IGF2BP2, 7 CDKAL1 and HHEX/IDE, 8 ADIPOQ, PPAR-γ, and RXR-α, 9 ABCC8 and KCNJ11, 10 TCF7L2, SLC30A8, PCSK1, and PCSK2 11 were involved in the T2DM. Recent studies have shown that signaling pathway, including PI3K/AKT-and AMPK signaling pathway, 12 mTOR signaling pathway, 13 insulin signaling pathway, 14 AGE/RAGE/JNK, STAT3/SCOS3, and RAS signaling pathway, 15 and ERK signaling pathway 16 were involved in progression of T2DM. Therefore, it is of great practical significance to explore the genes and signaling pathways of T2DM on islet cells.
RNA sequencing technology can rapidly detect gene expression on a global basis and are particularly useful in screening for differentially expressed genes (DEGs) in diseases. 17 RNA sequencing technology allows the investigation of gene expression in a high throughput manner with high sensitivity, specificity and repeatability. Significant amounts of data have been produced via the use of RNA sequencing and the majority of such data has been uploaded and stored in public databases. Indeed, some researchers found key genes and pathways in T2DM by integrated bioinformatics analysis.18-23 However, the comparative analysis of DEGs across a range of independent investigation might yield only a relatively limited amount of useful data with regard to T2DM advancement. The disadvantages of these single investigations might be overcome by NGS analysis, as this approach would make it possible to analyze the signaling pathways and interaction networks linked with the identified DEGs. This knowledge might help in elucidating the molecular mechanisms underlying T2DM and its associated complications.
In the present investigation, next generation sequencing (NGS) dataset was downloaded from the Gene Expression Omnibus (GEO) (GEO, http://www.ncbi.nlm.nih.gov/geo/) 24 : GSE81608. 25 DEGs were identified in T2DM. We then carried out gene ontology (GO), REACTOME pathway enrichment analysis, protein-protein interaction (PPI) network analysis, module analysis, miRNA-hub gene regulatory network, and TF-hub gene regulatory network analysis to elucidate the underlying molecular mechanisms. Finally, hub genes were validated by receiver operating characteristic curve (ROC). Collectively, the findings of the present investigation highlighted crucial genes and signaling pathways that might contribute to the pathology of T2DM and associated complications. The research flowchart of this investigation was shown in Figure 1. These may provide a basis for the advancement of future diagnostic, prognostic and therapeutic tools for T2DM.

Research design flow chart.
Materials and Methods
Data resources
NGS dataset GSE81608 25 was downloaded from the GEO database. The platform used for NGS data was the GPL16791 Illumina HiSeq 2500 (Homo sapiens). The GSE81608 dataset contained data from 1600 samples, including 949 T2DM samples (single human islet cells), and 651 healthy control samples (single human islet cells).
Identification of DEGs
Limma package in R software 26 is a tool to identify DEGs by comparing samples from GEO series. Limma package in R software was used to search for in messenger RNAs (mRNAs; DEGs) that were differentially expressed between T2DM and healthy control samples. The cutoff criteria were an adjusted P-value of <.05, whereas the logFC value were >0.181 for up regulated genes and <−0.27 for down regulated genes. DEG of this dataset was visualized with volcano map and hierarchical clustering heat map. The volcano plot was drawn using ggplot2 package in R software. Hierarchical clustering heat maps of DEG expression (up regulated genes and down regulated genes) were visualized with gplots package in R software.
GO and REACTOME pathway enrichment analysis of DEGs
GO enrichment analysis (http://geneontology.org/) 27 implements the annotation of biological processes (BP), cellular components (CC) and molecular functions (MF) of DEGs. REACTOME (https://reactome.org/) 28 is a database that stores large amounts of data on genomics, biological pathways, signaling pathways, diseases, drugs, and chemicals. The present investigation used Database for g:Profiler (http://biit.cs.ut.ee/gprofiler/) 29 to perform GO and REACTOME pathway enrichment analysis. P < .05 was considered to indicate a statistically significant difference.
Construction of the PPI network and module analysis
The IID interactome database (http://iid.ophid.utoronto.ca/) may be searched for associations between known and predicted proteins, and is commonly used to predict PPI information in molecular biology. 30 Cytoscape 3.8.2 (http://www.cytoscape.org/) 31 was used to visualize the results from the PPI network In this investigation, node degree, 32 betweenness centrality, 33 stress centrality, 34 and closeness centrality, 35 which constitutes a fundamental parameter in network theory, was adopted to evaluate the nodes in a network. The node degree betweenness centrality, stress centrality and closeness centrality methods were calculated using Cytoscape plugin Network Analyzer. Module analysis on the PPI network results was performed using the PEWCC1 36 clustering algorithm that comes with Cytoscape. Module analysis might be used to find out more connected gene groups. In addition, the module analysis were further analyzed for GO and pathway enrichment analysis.
MiRNA-hub gene regulatory network construction
Prediction of miRNA-hub genes was performed by miRNet database (https://www.mirnet.ca/). 37 According to the regulatory interaction, miRNA-hub gene regulatory network was constructed based on miRNet by Cytoscape 3.8.2 software. 31
TF-hub gene regulatory network construction
Prediction of TF-hub genes was performed by NetworkAnalyst database (https://www.networkanalyst.ca/). 38 According to the regulatory interaction, TF-hub gene regulatory network was constructed based on NetworkAnalyst by Cytoscape 3.8.2 software. 31
Validation of hub genes by receiver operating characteristic curve (ROC) analysis
ROC curve analysis was performed to evaluate the sensitivity and specificity of the hub genes for T2DM diagnosis using the R package “pROC.” 39 An area under the curve (AUC) value was determined and used to label the ROC effect. GEO datasets were used in ROC analysis. AUC > 0.8 indicated that the model had a good fitting effect. 40
Results
Identification of DEGs
A total of 927 genes were identified to be differentially expressed between T2DM and normal control samples with the threshold of adjusted P-value of <.05, and logFC value were >0.181 for up regulated genes and <−0.27 for down regulated genes. Among these DEGs, 461 were up regulated and 466 down regulated genes in T2DM compared with normal control samples and DEGs are listed in Supplemental Table S1. A heat map (Figure 2) and a volcano plot (Figure 3) for the identified DEGs was generated.

Heat map of differentially expressed genes. Legend on the top left indicate log fold change of genes. (A1-A651 = normal control samples; B1-B949 = T2DM samples).

Volcano plot of differentially expressed genes. Genes with a significant change of more than two-fold were selected. Green dot represented up regulated significant genes and red dot represented down regulated significant genes.
GO and REACTOME pathway enrichment analysis of DEGs
To identify the pathways which had the most significant involvement with the genes identified, up regulated and down regulated genes were submitted into g:Profiler for GO terms are listed in Supplemental Table S2 and REACTOME pathway enrichment analysis are listed in Supplemental Table S3. GO enrichment analysis revealed that in BP terms, the up regulated genes were mainly enriched in protein metabolic process and positive regulation of biological process. Down regulated genes were mainly enriched in establishment of localization and cellular metabolic process. In CC terms, up regulated genes were mainly enriched in intracellular anatomical structure and endomembrane system, whereas down regulated genes were mainly enriched in cytoplasm and intracellular anatomical structure. In MF terms, up regulated genes were mainly enriched in heterocyclic compound binding and protein binding, whereas down regulated genes were mainly enriched in catalytic activity and protein binding. REACTOME pathway enrichment analysis demonstrated that up regulated genes were significantly enriched in metabolism of proteins, and NR1H3 and NR1H2 regulate gene expression linked to cholesterol transport and efflux. Down regulated genes were significantly enriched in the metabolism and the citric acid (TCA) cycle and respiratory electron transport.
Construction of the PPI network and module analysis
Following the analysis based on the PPI networks, 4424 nodes and 8670 edges were identified in Cytoscape (Figure 4). The genes with higher scores were the hub genes, as the genes of higher node degree, betweenness centrality, stress centrality, and closeness centrality might be linked with T2DM. The top hub genes include APP, MYH9, TCTN2, USP7, SYNPO, GRB2, HSP90AB1, UBC, HSPA5, and SQSTM1, and topological properties of each hub genes in PPI network is given in Table 1. A total of 2 modules were selected through PEWCC1 analysis, and module 1 had nodes 98 and edges 117 (Figure 5A) and module 2 had nodes 81 and edges 248 (Figure 5B). Enrichment analysis demonstrated that modules 1 and 2 might be linked with RNA polymerase II transcription, intracellular anatomical structure, metabolism of proteins, protein metabolic process, positive regulation of biological process, metabolism, immune system, establishment of localization, cytoplasm, neutrophil degranulation, cellular metabolic process, intracellular anatomical structure, and protein binding.

PPI network of DEGs. The PPI network of DEGs was constructed using Cytoscap. Up regulated genes are marked in green; down regulated genes are marked in red. Big node represents nod with more number of interactions and small node represents nod with least number of interactions.
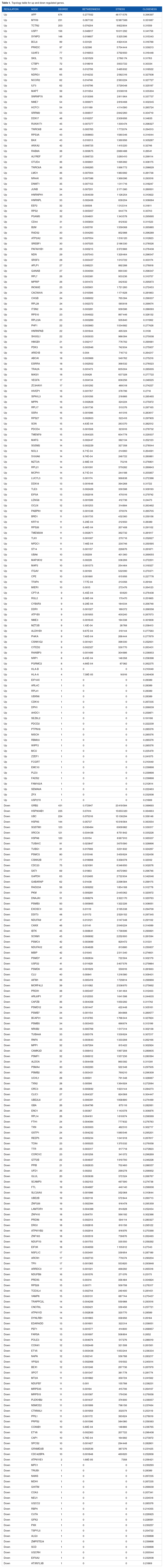
Topology table for up and down regulated genes.

Modules of isolated form PPI of DEGs: (A) the most significant module was obtained from PPI network with 98 nodes and 117 edges for up regulated genes and (B) the most significant module was obtained from PPI network with 81 nodes and 248 edges for down regulated genes. Up regulated genes are marked in green; down regulated genes are marked in red.
MiRNA-hub gene regulatory network construction
The hub genes of the DEGs in T2DM were performed by online databases miRNet. Based on the miRNAs, a miRNA-hub gene regulatory network was constructed with 2630 nodes (miRNA: 2345 and hub gene: 285) and 20 765 interaction pairs (Figure 6). PRKDC was the gene targets of 163 miRNAs (eg, hsa-mir-142-5p), MYH9 was the gene targets of 126 miRNAs (eg, hsa-mir-181b-3p), APP was the gene targets of 125 miRNAs (eg, hsa-mir-216b-5p), ILF3 was the gene targets of 107 miRNAs (eg, hsa-mir-3157-3p), SKIL was the gene targets 91 of miRNAs (eg, hsa-mir-1294), HSPA8 was the gene targets of 116 of miRNAs (eg, hsa-mir-3661), HSP90AB1 was the gene targets of 103 of miRNAs (eg, hsa-mir-200a-3p), SQSTM1 was the gene targets of 94 of miRNAs (eg, hsa-mir-520d-5p), HSPA5 was the gene targets of 88 of miRNAs (eg, hsa-mir-573), and GRB2 was the gene targets of 65 of miRNAs (eg, hsa-mir-1291), and topological properties of each hub genes and miRNAs in miRNA-hub gene regulatory network are listed in Table 2.

MiRNA-hub gene regulatory network. The purple color diamond nodes represent the key miRNAs; up regulated genes are marked in green; down regulated genes are marked in orange.
miRNA-target gene and TF-target gene interaction.

TF-hub gene regulatory network construction
The hub genes of the DEGs in T2DM were performed by online databases NetworkAnalyst. Based on the TFs, a TF-hub gene regulatory network was constructed with 477 nodes (TF: 192 and hub gene: 285) and 8507 interaction pairs (Figure 7). BCL6 was the gene targets of 60 TFs (eg, NOTCH1), MYH9 was the gene targets of 53 TFs (eg, PPARD), NCOR2 was the gene targets of 50 TFs (eg, HIF1A), APP was the gene targets of 45 TFs (eg, SMARCA4), NDRG1 was the gene targets of 44 TFs (eg, SUZ12), UBC was the gene targets of 64 TFs (eg, TAF7L), HSP90AB1 was the gene targets of 49 TFs (eg, RUNX2), TUBA1C was the gene targets of 47 TFs (eg, MITF), HSPA5 was the gene targets of 44 TFs (eg, YAP1), and HSPA8 was the gene targets of 39 TFs (eg, E2F1), and topological properties of each hub genes and TFs in TF-hub gene regulatory network are listed in Table 2.

TF-hub gene regulatory network. The blue color triangle nodes represent the key TFs; up regulated genes are marked in green; down regulated genes are marked in red.
Validation of hub genes by receiver operating characteristic curve (ROC) analysis
Validated by ROC curves, we found that 10 hub genes had high sensitivity and specificity, including APP (AUC = 0.853), MYH9 (AUC = 0.852), TCTN2 (AUC = 0.881), USP7 (AUC = 0.862), SYNPO (AUC = 0.893), GRB2 (AUC = 0.850), HSP90AB1 (AUC = 0.870), UBC (AUC = 0.865), HSPA5 (AUC = 0.902), and SQSTM1 (AUC = 0.875) (Figure 8). The hub genes might be biomarkers of T2DM and have positive implications for early medical intervention of the disease.

ROC curve validated the sensitivity, specificity of hub genes as a predictive biomarker for T2DM: (A) APP, (B) MYH9, (C) TCTN2, (D) USP7, (E) SYNPO, (F) GRB2, (G) HSP90AB1, (H) UBC, (I) HSPA5, and (J) SQSTM1.
Discussion
Although there are various investigations on T2DM that have been conducted, the mortality of T2DM is still high. This might be due to the lack of valid biomarkers for detection of early stage T2DM and of valid treatment for T2DM. Therefore, molecular mechanisms of T2DM are necessary for scientists to find the treat and diagnosis method of T2DM. Because of the fast advancement of NGS technology, it is more convenient to find out the genetic modification of development of diseases. NGS facilitates us to examine the gene, the genetic modification in T2DM, which had been proved to be a better approach to find novel biomarkers in other metabolic diseases.
In the present investigation, we observed whether there were more beneficial genes which could be better biomarkers for the diagnosis, prognosis and therapeutic for T2DM. In order to find out the significant gene of T2DM, we analyzed the NGS data GSE81608 in Limma, where a total number of 927 DEGs were obtained between T2DM and normal control, comprising was 461 up regulated and 466 down regulated genes. CTBP1 41 and TRNC 42 are involved in the pathogenesis of T2DM. A previous study has demonstrated that SST (somatostatin) serves an essential role in obesity. 43 Therefore, the data suggest that the identified DEGs might participant in the development of T2DM and associated complications and contribute to T2DM treatment.
Then, databases including GO and REACTOME were selected to do gene enrichment analysis. Metabolism of proteins, 44 metabolism, 45 the citric acid (TCA) cycle and respiratory electron transport, 46 gluconeogenesis, 47 immune system, 48 heterocyclic compound binding, 49 protein binding, 50 establishment of localization, 51 cellular metabolic process, 52 cytoplasm, 53 and catalytic activity 54 were the GO terms and signaling pathways responsible for the advancement of T2DM. A previous study showed that IGFBP2, 55 APOH (apolipoprotein H), 56 ANXA2, 57 BAX (BCL2 associated X, apoptosis regulator), 58 PCSK1N, 59 PDK4, 60 CPE (carboxypeptidase E), 61 OCLN (occludin), 62 CD44, 63 NDN (necdin, MAGE family member), 64 MLXIPL (MLX interacting protein like), 65 CD36, 66 SREBF1, 67 NR4A1, 68 PCSK2, 69 CHGB (chromogranin B), 70 PDK3, 71 PDCD4, 72 EIF5A, 73 NRP1, 74 ABCA1, 75 DNMT1, 76 MYH9, 77 HMGB1, 78 B4GALT5, 79 B2M, 80 MAP3K12, 81 KSR2, 82 NPY (neuropeptide Y), 83 CHGA (chromogranin A), 84 CD47, 85 DLK1, 86 PDK4, 87 CPE (carboxypeptidase E), 61 OCLN (occludin), 62 CXXC4, 88 PEMT (phosphatidylethanolamine N-methyltransferase), 89 FADS2, 90 RREB1, 91 HNRNPAB (heterogeneous nuclear ribonucleoprotein A/B), 92 CPT1A, 93 ALDH1B1, 94 ESRRA (estrogen related receptor alpha), 95 NISCH (nischarin), 96 SSTR3, 97 ND1, 98 NCOR2, 99 RBP4, 100 GSTP1, 101 CYB5A, 102 G6PC2, 103 DNAJC15, 104 TMED6, 105 PSMD6, 106 CLU (clusterin), 107 TTR (transthyretin), 108 TXN (thioredoxin), 109 LAMTOR1, 110 GLUL (glutamate-ammonia ligase), 111 NEU1, 112 HSPA8, 113 AP3S2, 114 COX4I1, 115 MT2A 116 MTCH2, 117 ESD (esterase D), 118 UBE2L6, 119 SCD (stearoyl-CoA desaturase), 120 MGST3, 121 NQO1, 122 NSMCE2, 123 and PRSS1 124 played an important role in T2DM. Quintela et al, 125 Yuan et al, 126 Cacace et al, 127 Hao et al, 128 Beckelman et al, 129 Liu et al, 130 Sekiguchi et al, 131 Castillon et al, 132 O’Donnell-Luria et al, 133 Coupland et al, 134 Koufaris et al, 135 Qvist et al, 136 Richter et al, 137 Torres et al, 138 Jeong et al, 139 Bermejo-Bescós et al, 140 Ramon-Duaso et al, 141 Guilarte, 142 Mukaetova-Ladinska et al, 143 Fazeli et al, 144 Butler et al, 145 Nackenoff et al, 146 Konyukh et al, 147 Hu et al, 148 Kaur et al, 149 Nakamura et al, 150 Liu et al, 151 Obara et al, 152 Herrmann et al, 153 Ozgen et al, 154 Masciullo et al, 155 Perrone et al, 156 Su et al, 157 Zhao et al, 158 Iqbal et al, 159 Gal et al, 160 Wang et al, 161 Stefanović et al, 162 Zahola et al, 163 Bik-Multanowski et al, 164 Mata et al, 165 Li et al, 166 Payton et al, 167 and Chai et al 168 indicated that UBA6, TIA1, DPP6, USP7, EEF2, ITM2B, DPH1, PAK3, KMT2E, MAPT (microtubule associated protein tau), HCFC1, BRD1, TAOK2, PHF1, STMN2, APP (amyloid beta precursor protein), MBNL2, APLP1, MAP2, SRRM2 CST3, SRRM2, CST3, PLD3, SEZ6L2, DOC2A, PI4KA, GNAO1, TRA2A, MIDN (midnolin), HOOK3, MCPH1, SACS (sacsin molecular chaperone), TUBA4A, ASAH1, ATP6V1B2, SVBP (small vasohibin binding protein), AIFM1, UBC (ubiquitin C), IFI30, SCGN (secretagogin, EF-hand calcium binding protein), MTRNR2L12, GBA (glucosylceramidase beta), TXN2, NQO2, and PPIL1 were involved in the development and progression of cognitive impairment. RPS3A, 169 PGAM5, 170 RPL7, 171 TLK1, 172 DDR1, 173 ILF3, 174 TNRC6A, 175 GGCX (gamma-glutamyl carboxylase), 176 S100A6, 177 LSAMP (limbic system associated membrane protein), 178 KCNA5, 179 LUC7L3, 180 ATAD3C, 181 SRSF3, 182 MCU (mitochondrial calcium uniporter), 183 ATP2A2, 184 GAA (glucosidase alpha, acid), 185 MAGI1, 186 WIPF2, 187 VAMP8, 188 UCHL1, 189 CLIC1, 190 PSMB5, 191 GRB2, 192 MPSTE24, 193 COX6B1, 194 SQSTM1 195 , COTL1 196 , CD63 197 , NDUFB7 198 , BEX1 199 and MTRNR2L8 200 plays a major role in mediating cardiovascular diseases progression. HLA-A, 201 VEGFA (vascular endothelial growth factor A), 202 RPS26, 203 BMP6, 204 HLA-B, 205 IER3IP1, 206 MT1E, 207 ACADM (acyl-CoA dehydrogenase medium chain), 208 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) 209 are associated with progression of Type 1 diabetes mellitus. PEMT (phosphatidylethanolamine N-methyltransferase), 210 INSM1, 211 BCL6, 212 RUNX1T1, 213 PGRMC2, 214 ARID1B, 215 CITED2, 216 KLF13, 217 PPT1, 218 ARRDC3, 219 HSPA5, 220 MDH2, 221 and COA3, 222 have been previously reported to be a key biomarkers for the early detection of obesity. A previous study demonstrated that IGFBP5, 223 PRDX6, 224 PKM (pyruvate kinase M1/2), 225 PRDX1, 226 and USP22 227 were more highly expressed in diabetic nephropathy. Durgin et al, 228 Zhang et al, 229 Hamada et al, 230 Gong et al, 231 Li et al, 232 Lin et al, 233 and Schweigert et al 234 suggested that CYB5R3, CACNA1A, GLCCI1, CAP1, HSP90AB1, BLVRA (biliverdinreductase A), and CRIP1 were involved in the progression of hypertension. These results suggested that these, cognitive impairment, cardiovascular diseases, obesity, diabetic nephropathy and hypertension responsible genes might influence the development of T2DM through the altered expression. Therefore, these genes might involve in these GO terms and pathways are most likely to be important in the development of T2DM and T2DM associated complications.
By PPI network and module analysis, we identified the hub genes that might affect the origin or advancement of T2DM. TCTN2, SYNPO (synaptopodin), PSMD12, PSMC4, TUBA1C, PSMC5, PSMD7, and RAD23A might serve as a novel target for early diagnosis and specific therapy of T2DM and T2DM associated complications, and the related mechanisms need to be further investigation.
In addition, miRNA-hub gene regulatory network construction and TF-hub gene regulatory network were constructed. In addition, miRNA-mRNA networks were constructed. The roles of hub genes, miRNA and TF in the pathogenesis of T2DM are discussed. Hsa-mir-142-5p, 235 hsa-mir-1291, 236 NOTCH1, 237 PPARD (peroxisome proliferator-activated receptor delta), 238 HIF1A, 239 RUNX2, 240 and E2F1 241 levels are correlated with disease severity in patients with T2DM. Previous studies have demonstrated that hsa-mir-216b-5p 242 and hsa-mir-200a-3p 243 appears to be expressed in Type 1 diabetes. Hsa-mir-1294, 244 SUZ12, 245 and YAP1 246 were responsible for progression of cognitive impairment. Hsa-mir-573 247 and SMARCA4 248 were linked with progression of hypertension. Therefore, cognitive impairment and hypertension responsible biomarkers might be used as a diagnostic biomarker because of its essential role in the pathogenesis in early T2DM. Our study also suggests that PRKDC (protein kinase, DNA-activated, catalytic subunit), SKIL (SKI like proto-oncogene), NDRG1, hsa-mir-181b-3p, hsa-mir-3157-3p, hsa-mir-3661, hsa-mir-520d-5p, TAF7L, and MITF (Microphthalmia-associated transcription factor) are the novel biomarkers of the entire process of T2DM and T2DM associated complications development and might be used as the novel diagnostic biomarker for T2DM and T2DM associated complications.
The conduct of updating methods was calculated the classification work in which collected a higher score in efficiency, AUC, specificity, and sensitivity. As a result, 10 hub genes with AUC > 0.80 showed excellent diagnostic value for T2DM, and thus were considered as hub genes of T2DM, including APP, MYH9, TCTN2, USP7, SYNPO, GRB2, HSP90AB1, UBC, HSPA5, and SQSTM1. Through these analyses, we expect to provide novel insights into the molecular pathogenesis of T2DM and its associated complications and provide a more detailed molecular mechanism for the development of T2DM treatment. Although bioinformatics analysis has been performed in these present investigations, some limitations exist. Lacking of experimental validation of hub genes is a limitation of the study. In addition, we do not conduct in vitro and in vivo experiments of hub genes in T2DM. Corresponding experiments will be performed to verify in our future investigation, thus conversely testifying in bioinformatics analysis.
In conclusion, the present study identified 10 hub genes (APP, MYH9, TCTN2, USP7, SYNPO, GRB2, HSP90AB1, UBC, HSPA5, and SQSTM1) with crucial role in progression of T2DM; our results suggested these genes could add a new dimension to our understanding of the T2DM and might be served as potential biomarkers that will be assisting endocrinologist in developing novel therapeutic strategies for T2DM patients. However, there are some limitations in this study. Further larger clinical sample size and in-depth clinical experiments are needed to clarify the clear mechanism and warrant the prognostic value of these DEGs in T2DM.
Supplemental Material
sj-docx-1-end-10.1177_11795514231155635 – Supplemental material for Bioinformatics Analysis of Next Generation Sequencing Data Identifies Molecular Biomarkers Associated With Type 2 Diabetes Mellitus
Supplemental material, sj-docx-1-end-10.1177_11795514231155635 for Bioinformatics Analysis of Next Generation Sequencing Data Identifies Molecular Biomarkers Associated With Type 2 Diabetes Mellitus by Varun Alur, Varshita Raju, Basavaraj Vastrad, Chanabasayya Vastrad, Satish Kavatagimath and Shivakumar Kotturshetti in Clinical Medicine Insights: Endocrinology and Diabetes
Supplemental Material
sj-docx-2-end-10.1177_11795514231155635 – Supplemental material for Bioinformatics Analysis of Next Generation Sequencing Data Identifies Molecular Biomarkers Associated With Type 2 Diabetes Mellitus
Supplemental material, sj-docx-2-end-10.1177_11795514231155635 for Bioinformatics Analysis of Next Generation Sequencing Data Identifies Molecular Biomarkers Associated With Type 2 Diabetes Mellitus by Varun Alur, Varshita Raju, Basavaraj Vastrad, Chanabasayya Vastrad, Satish Kavatagimath and Shivakumar Kotturshetti in Clinical Medicine Insights: Endocrinology and Diabetes
Supplemental Material
sj-docx-3-end-10.1177_11795514231155635 – Supplemental material for Bioinformatics Analysis of Next Generation Sequencing Data Identifies Molecular Biomarkers Associated With Type 2 Diabetes Mellitus
Supplemental material, sj-docx-3-end-10.1177_11795514231155635 for Bioinformatics Analysis of Next Generation Sequencing Data Identifies Molecular Biomarkers Associated With Type 2 Diabetes Mellitus by Varun Alur, Varshita Raju, Basavaraj Vastrad, Chanabasayya Vastrad, Satish Kavatagimath and Shivakumar Kotturshetti in Clinical Medicine Insights: Endocrinology and Diabetes
Footnotes
Acknowledgements
I thank Yurong Xin, Regeneron Pharmaceuticals, Inc., Tarrytown, New York, USA, very much, the author who deposited their NGS dataset GSE81608, into the public GEO database.
Declarations
Informed consent
No informed consent because this study does not contain human or animals participants.
Ethics approval
Not applicable.
Trial registration
Not applicable.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.