
Abstract
Background:
Cutaneous Rosai–Dorfman disease (CRDD) is a rare extranodal histiocytosis that can mimic other dermatoses, particularly when hallmark emperipolesis is absent.
Case Presentation:
A 50-year-old man presented with 5 months of intensely pruritic lesions affecting the palms, soles, flexures, and limbs. Skin biopsy revealed a nodular infiltrate of foamy histiocytes extending into the subcutis. Immunohistochemistry showed S100⁺, CD68⁺, CD1a⁻, while emperipolesis was not identified. Systemic evaluation was unremarkable. He was treated with oral prednisone, which was discontinued due to steroid-induced hyperglycemia, and methotrexate 15 mg/week, achieving approximately 60% improvement of symptoms at 6 months.
Conclusion:
CRDD may present atypically, and the absence of emperipolesis does not exclude the diagnosis. Careful clinicopathologic correlation and immunohistochemical analysis are essential for accurate diagnosis and guiding individualized therapy.
Keywords
Background
Rosai–Dorfman disease (RDD), originally termed sinus histiocytosis with massive lymphadenopathy, is a rare benign proliferative disorder of non–Langerhans histiocytes. It was first described by Destombes in 1965 and later characterized as a distinct clinicopathological entity by Rosai and Dorfman in 1969. RDD typically involves overproduction and accumulation of S-100⁺/CD68⁺ histiocytes within lymph nodes and extranodal tissues. Most patients are children or young adults (peak incidence in the second–third decades). The disease shows a male predominance and is significantly more common in people of African descent; it is reported less frequently in Asians.1-3
The classic clinical presentation is painless, massive bilateral cervical lymphadenopathy. Patients often have systemic symptoms (eg, low-grade fever) and laboratory abnormalities. Common findings include normocytic anemia, neutrophilic leukocytosis, an elevated erythrocyte sedimentation rate (ESR), and polyclonal hypergammaglobulinemia. Histopathology of an involved lymph node shows massively dilated sinuses filled with large histiocytes exhibiting emperipolesis (intact lymphocytes or plasma cells within histiocyte cytoplasm). 1
Extranodal involvement occurs in roughly 40% of RDD cases. The most common extranodal sites are the skin and soft tissues, orbit, central nervous system, and upper respiratory or gastrointestinal tracts. The skin is the single most frequent extranodal organ involved. By contrast, purely cutaneous RDD without any nodal disease is exceedingly rare.1-4 Herein, we present a rare case of cutaneous Rosai–Dorfman disease in a 50-year-old male with pruritic palmoplantar and flexural lesions.
Case Presentation
A 50-year-old man presented to our dermatology clinic with a 5-month history of intensely pruritic and mildly burning skin lesions affecting the upper and lower extremities, including the palms, soles, and flexural regions. The scalp, face, nails, and mucous membranes were unaffected. The eruption initially appeared on the lower extremities and gradually spread over time (Figure 1).

Clinical presentation of cutaneous lesions.
General physical examination and systemic review revealed no lymphadenopathy, fever, weight loss, night sweats, or gastrointestinal symptoms. Baseline laboratory, inflammatory, and serologic investigations are summarized in Table 1 and were within normal reference ranges. However, serum protein electrophoresis revealed polyclonal hypergammaglobulinemia, with elevated gamma globulins (18.5 g/l, 23.8%), while the remaining protein fractions were within normal limits (Figure 2).
Baseline Laboratory, Inflammatory, and Serologic Investigations.


Serum protein electrophoresis demonstrating a polyclonal increase in gamma globulins.
Six months prior to presentation, roughly 1 month before the onset of his skin lesions, the patient had an acute myocardial infarction. He subsequently underwent coronary angiography with placement of 2 drug-eluting stents. Post-procedure, he was started on aspirin 81 mg daily, prasugrel 10 mg daily, and atorvastatin 40 mg daily, which he has continued without interruption (Table 2).
Differential Diagnoses of Cutaneous Rosai–Dorfman Disease: Key Histopathologic and Immunophenotypic Features.

The bold text indicates clinically significant findings relevant to this case report.
Histopathological examination of the skin biopsy, taken from a 15 mm × 10 mm papule on the trunk, revealed a dense dermal and subcutaneous infiltrate composed predominantly of large histiocytic cells, including foamy histiocytes and xanthoma cells, admixed with lymphocytes and plasma cells, without cytologic atypia (Figure 3). Routine evaluation was performed using hematoxylin and eosin (H&E) staining. Immunohistochemical analysis demonstrated positivity for CD68 and S-100 in the lesional histiocytes, with negative CD1a staining, confirming the diagnosis of cutaneous Rosai–Dorfman disease (CRDD; Figure 4). Immunohistochemical staining was validated using appropriate positive controls: palatine tonsil tissue served as an external positive control for CD68, while native skin structures within the biopsy specimen were used as internal positive controls for CD1a and S-100. CD207 (langerin) immunostaining was not available in our pathology laboratory. Histopathological images were acquired using a Nikon microscope that was not equipped with an integrated scale bar feature. The patient was referred to hematology for multidisciplinary care.
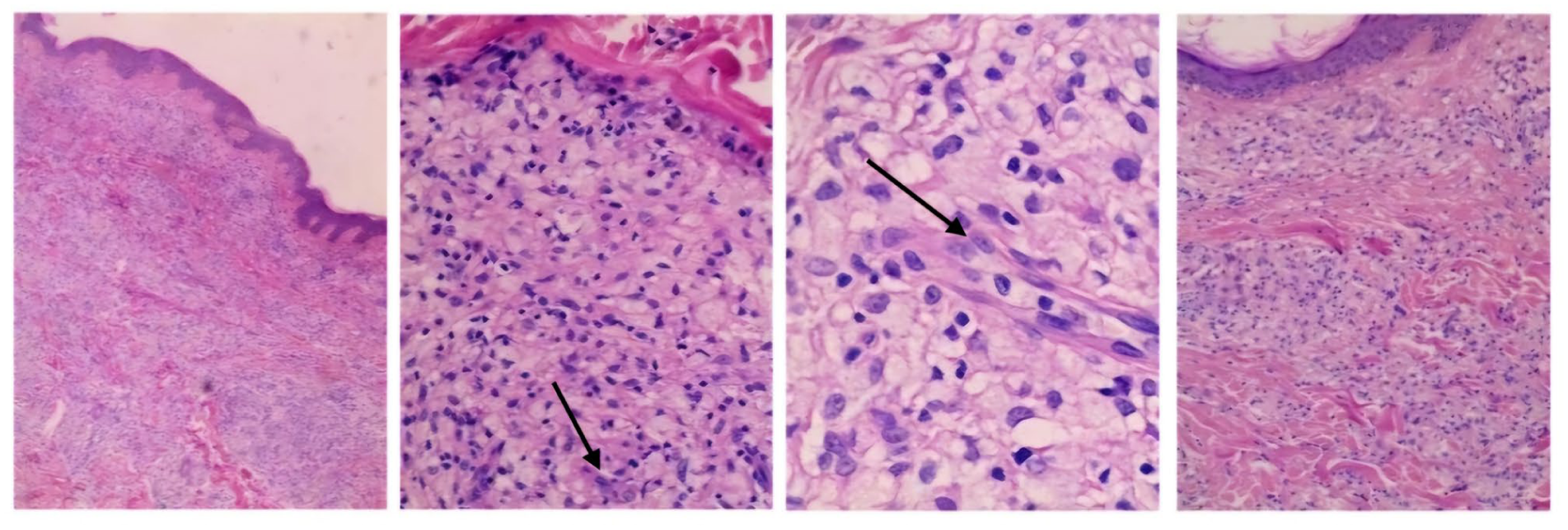
Dermal accumulation of histiocytes with enlarged round to oval hypochromatic nuclei and abundant clear to eosinophilic cytoplasm, admixed with scattered plasma cells and lymphocytes (H&E, ×100).

Immunohistochemical staining showing positivity for CD68 (A) and S-100 (B), with negativity for CD1a (C), supporting the diagnosis.
special histochemical stains, including Ziehl–Neelsen, periodic acid–Schiff, and Grocott methenamine silver, were performed on the biopsy specimen and were negative for mycobacterial and fungal organisms. Tissue cultures for bacterial, mycobacterial, and fungal pathogens were also negative. Systemic evaluation, including lymph node examination and imaging studies, revealed no evidence of nodal or extracutaneous involvement.
The patient received systemic therapy for a total of 6 months. Oral prednisone (30 mg/day) was administered for the first 4 months, together with weekly methotrexate (15 mg/week) and folic acid supplementation, along with gastric protection. During corticosteroid therapy, the patient was monitored clinically and biochemically, with follow-up focusing on lesion progression, pruritus severity, and laboratory parameters. At month four, the patient developed steroid-induced hyperglycemia (fasting glucose 168 mg/dl) with blurred vision, prompting gradual tapering and discontinuation of prednisone. Fasting glucose subsequently normalized (94 mg/dl) without antidiabetic therapy. Methotrexate was continued as monotherapy for the following 2 months, with ongoing clinical assessment and routine laboratory monitoring (complete blood count and liver function tests). At 6-month follow-up, the patient reported approximately 60% improvement in pruritus, with near-complete resolution of smaller papules, while larger plaques persisted (Figure 5).

Clinical improvement after 6 months of therapy with methotrexate.
Discussion
RDD is a rare histiocytic disorder that typically presents with massive, painless lymphadenopathy in the head and neck with general symptoms, while involvement of other organ systems is less common. Isolated CRDD is exceedingly rare, representing only about 3% of all RDD cases.1,5,6 CRDD is a distinct clinicopathologic variant of Rosai–Dorfman disease limited to the skin. It remains localized to the dermis and subcutis without lymph nodes or systemic involvement. CRDD typically presents in middle-aged adults, with a median age of about 43 to 45 years, and shows a clear female predominance. Clinically, patients develop painless papules, nodules, or plaques that are often yellowish or dark red, which may be solitary or multifocal and occur most frequently on the face, trunk, and extremities. Unlike systemic disease, purely cutaneous cases lack lymphadenopathy, organ involvement, or serologic abnormalities. Although its histologic features, such as S100⁺ histiocytes with emperipolesis, resemble systemic Rosai–Dorfman disease, CRDD differs in its epidemiology, clinical presentation, and course, supporting its recognition as a separate clinical entity.5-7 In our case, the dermal biopsy showed a dense infiltrate of foamy (lipidized) histiocytes admixed with lymphocytes and plasma cells, without identifiable emperipolesis. Although emperipolesis is considered a hallmark of Rosai–Dorfman disease, it may be scant or absent in cutaneous and extranodal forms, particularly in longstanding lesions, as reported in previous studies. Such a prominent xanthomatous pattern is uncommon in CRDD and represents an unusual morphologic variant that may complicate recognition without careful histologic and immunohistochemical analysis.8-14
The patient was 50 years old, which is older than the usual age range for CRDD, as the disease most commonly affects middle-aged women. Lesions were intensely pruritic and involved acral and flexural sites, including the palms and soles—locations rarely affected. Severe pruritus, though uncommon, has been reported in isolated cases. These features underscore that CRDD may rarely present with itch and palmoplantar involvement.5-7 CRDD most often involves the trunk and limbs. In a review of 53 cases, Mohaghegh et al found lesions mainly on the trunk (52%) and extremities (43%), while acral and flexural sites are usually spared. Involvement of the palms, soles, and flexures is rare, with only isolated reports of painful multifocal palmoplantar lesions. In our patient, the rash extensively involved acral sites (palms and soles) and flexures which is a pattern only infrequently reported in CRDD. 6
The main differential diagnoses included non-Langerhans cell histiocytosis, Langerhans cell histiocytosis, cutaneous lymphoma, sarcoidosis, and infectious granulomatous diseases. The strong and diffuse S-100 positivity in lesional histiocytes ruled out histiocytic disorders that typically lack this marker, while clinical, histopathologic, and immunohistochemical findings excluded the other entities (Table 1).9,10 Infectious granulomatous diseases, including mycobacterial and deep fungal infections, were excluded by the absence of necrotizing granulomas on histology and by negative special stains (Ziehl–Neelsen, PAS, and Grocott methenamine silver), as well as negative tissue cultures. Cutaneous lymphoma was considered unlikely given the lack of cytologic atypia, absence of a monomorphic lymphoid infiltrate, and the presence of a predominant population of S100⁺/CD68⁺ histiocytes rather than lymphoid cells. In addition, comprehensive clinical evaluation, including lymph node examination and imaging studies, revealed no evidence of nodal or systemic involvement, supporting the diagnosis of purely cutaneous Rosai–Dorfman disease. 11
The absence of pathognomonic emperipolesis in skin lesions can profoundly delay diagnosis. In long-standing or purely CRDD, fibrosis and chronic inflammation often predominate, rendering histiocytes less conspicuous and emperipolesis scarce. Indeed, extranodal CRDD is known to exhibit infrequent typical histiocytes and reduced emperipolesis compared to nodal disease. 12 A large review of CRDD cases found that classic features (large S100^ + foamy histiocytes with engulfed lymphocytes) are sometimes inconspicuous, highlighting that pathologists must rely on the overall immunophenotype (S100+/CD68+/CD1a) when hallmark emperipolesis is absent. 13 This need for careful histopathologic judgment often means that CRDD is misinterpreted initially, especially when no lymphadenopathy or systemic “B” symptoms prompt consideration of Rosai–Dorfman. As Gameiro et al emphasized, awareness of stage-dependent histologic variation (with older lesions lacking classical signs) is crucial to avoid misdiagnosis. 7 In a resource-limited setting without advanced molecular tests, therefore, the expertise of an experienced dermatopathologist is invaluable to recognize these subtle clues and initiate appropriate further workup. 14
The treatment of CRDD is not standardized. Many localized CRDD lesions are observed or treated with local therapies (topical/intralesional steroids, surgery). In more extensive or symptomatic cases, systemic therapies are used. Reported regimens include corticosteroids, methotrexate, thalidomide, isotretinoin, dapsone, or cytotoxic agents. In our patient’s low-resource setting, after biopsy diagnosis he was treated initially with oral prednisone, with partial response. Unfortunately, corticosteroids caused hyperglycemia and visual blurring, necessitating withdrawal. Methotrexate (15 mg/week) was continued as a steroid-sparing agent (with folate), which is reasonable based on published cases. At 6 months follow-up, the patient’s pruritus had improved (~60%) and small papules resolved, though larger plaques persisted – illustrating that CRDD often has an indolent course and may improve slowly. Ultimately, complete remission or long-term control is the goal, and additional options (eg, immunomodulators or targeted therapy) could be considered if needed, per recent consensus guidelines.6,15
A literature review was conducted to identify peer-reviewed case reports and case series describing adult patients with purely CRDD. An electronic search of PubMed/MEDLINE was performed for articles published in English from January 2000 to December 2025 using combinations of the terms “cutaneous Rosai–Dorfman disease,” “Rosai–Dorfman disease,” “sinus histiocytosis,” “cutaneous histiocytosis,” and “case report” or “case series.” Reference lists of relevant articles were manually screened to identify additional eligible reports. Inclusion criteria were: (1) adult patients (⩾18 years); (2) histologically confirmed CRDD confined to the skin without nodal or systemic involvement; (3) immunohistochemical confirmation showing S100 positivity with CD68 and/or CD163 positivity and CD1a negativity; and (4) adequate clinical and histopathologic description. Exclusion criteria included pediatric cases, systemic or mixed cutaneous–systemic disease, other histiocytic disorders, reports lacking immunohistochemical confirmation, review articles, abstracts, non–peer-reviewed publications, and duplicate cases. Data were extracted independently from each eligible report, including patient demographics, lesion distribution, presence of pruritus, histologic evidence of emperipolesis, immunophenotype, treatment modality, and clinical outcome. Given the descriptive nature of available data, findings were synthesized qualitatively and summarized in Table 3.6,16-19
Clinicopathologic Characteristics, Immunophenotype, Treatment, and Outcomes of Reported Adult Cases of Purely Cutaneous Rosai–Dorfman Disease Lacking Emperipolesis.

The bold text indicates clinically significant findings relevant to this case report.
The reported CRDD cases share several consistent patterns. Patients are predominantly middle-aged (average in the 40s) with a clear female bias. Lesions most often arise on the face, trunk, or extremities; involvement of palms, soles or flexural areas is uncommon. Most patients report no symptoms from the lesions, and itching or pain is rarely noted. Histopathology uniformly shows a dense dermal/subcutaneous infiltrate of lipid-laden histiocytes intermixed with plasma cells and lymphocytes. The pathognomonic emperipolesis (engulfed lymphocytes) is typically seen, although it may be subtle in chronic lesions. Immunophenotyping is key: all cases are S-100⁺, CD68⁺/CD163⁺ and CD1a⁻, distinguishing CRDD from other histiocytoses. Treatments have varied widely, reflecting lack of guidelines: common interventions include surgical excision (especially for solitary nodules) and corticosteroids (topical, intralesional or systemic). Many patients improved with therapy; for example, surgical removal led to resolution in 1 series, and intralesional steroids yielded marked clearing in others. Some cases (as in Zhang et al ) even regressed without treatment, underscoring the often-indolent course. There is no standard therapy, but most reports describe excision or corticosteroids as first-line. Because CRDD often follows a benign, slowly resolving course, many recommend conservative management unless lesions are disfiguring or symptomatic. For refractory or multisite disease, systemic treatments (eg, methotrexate, immunomodulators or targeted agents) have been tried with variable success.6,16-19
This report has several limitations inherent to single-patient case studies. Findings from 1 patient cannot be generalized to all cases of cutaneous Rosai–Dorfman disease. Some diagnostic markers, such as CD163 or langerin (CD207), and molecular testing were not available in our laboratory, which limited further characterization of the lesion. Despite these limitations, this case highlights an atypical presentation of CRDD and emphasizes the importance of clinicopathologic correlation when emperipolesis is absent.
Conclusion
This case emphasizes that cutaneous RDD, though rare, can present atypically with unusual patient demographics, uncommon lesion locations, and significant pruritus. Emperipolesis is a characteristic feature of RDD, but it may be absent in cutaneous forms, particularly in longstanding or fibrotic lesions. Its absence should not exclude the diagnosis if other histopathologic and immunohistochemical findings, notably an S100⁺/CD68⁺ and CD1a⁻ profile, are supportive. Careful clinico-pathological correlation is therefore essential, especially in low-resource settings where advanced testing is limited. Greater awareness of such atypical presentations can help avoid misdiagnosis and ensure appropriate management of CRDD.
Footnotes
Ethical Considerations
Ethical approval was not required for this case report, as it involves a single patient and does not meet the criteria for research requiring institutional review board (IRB) oversight. The use of authorized medications was consistent with clinical practice guidelines, and the management of this individual patient did not require IRB approval.
Consent to Participate
Written informed consent was obtained from the patient prior to publication of this case report and any accompanying images. This consent includes permission for the use of unauthorized, off-label medications as part of the treatment plan. The completed consent form is available to the Editor upon request and will be treated confidentially.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and accompanying clinical images.
Author Contributions
Lina Al-Soufi, MD: Conception of the case report, clinical diagnosis, patient management, data collection, and critical revision of the manuscript.
Aya Marashli, MD: Drafted the manuscript, performed literature review, coordinated manuscript preparation, and served as corresponding author.
Razan Younis, MD: Contributed to clinical data interpretation, follow-up of the patient, and review of the dermatologic findings.
Areeb Saeed Alkasem, MD: Assisted in histopathologic data analysis, literature review, and manuscript editing.
Firas Hussein, MD: Provided oncologic consultation, contributed to diagnostic interpretation, and revised the final manuscript for intellectual content.
Zuheir Al-Shehabi, MD: Performed histopathologic examination, immunohistochemical interpretation, and contributed to diagnostic confirmation.
All authors reviewed and approved the final version of the manuscript and agreed to be accountable for all aspects of the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data pertinent to this case report have been included in this article. Further inquiries can be directed to the corresponding author.