
Abstract
Rosai-Dorfman disease (RDD) is a clonal histiocytic proliferation characterized by large S100 positive histiocytes with variable emperipolesis. Extranodal locations were confirmed with the central nervous system or the meninges involvement in less than 5% of cases, which is marked as a significant differential diagnosis of meningiomas in radiological and intra-operative pathological examination. Histopathology and immunohistochemistry are the keys to definitive diagnosis. We present a case of bifocal Rosai-Dorfman disease in a 26-year-old man, mimicking Lymphoplasmacyte-rich Meningioma. This case allows us to demonstrate the diagnosis pitfalls encountered in this localization.
Introduction
Rosai-Dorfman disease is actually defined by the World Health Organization (WHO) as a clonal histiocytic proliferation. It is composed of a mixed inflammatory infiltrate including emperipolesis with histiocytic engulfment of intact inflammatory cells. This feature may be inconspicuous or absent. 1 Extranodal involvement is documented in 43% of the patients. Primary central nervous involvement is extremely rare (less than 5%);2,3 in such location a wide morphologic spectrum is encountered. This complicates the diagnosis and delays the therapeutic management.
Herein, we report an illustrative case of bifocal Rosai-Dorfman disease in a 26-year-old man mimicking lymphoplasmacyte-rich meningioma to propose useful morphological features to help ruling out the differential diagnosis.
Clinical Presentation
We report the case of 26-year-old man without any previous medical history. He suffered of vomiting, headaches, and vertigo during 2 months. He also reported a progressive deterioration of visual acuity. Clinical examination was normal and there was no lymphadenopathy.
Non-contrast Computed Tomography (CT) revealed bifocal hyperdense and compressive extra-axial masses with perilesional edema in the right parieto-occipital region and the left frontal region. These masses produce a mass effect on the lateral ventricles and the middle line (Figure 1). At this stage, Meningiomas and Meningeal metastases are the main differential radiological diagnosis that we discussed.
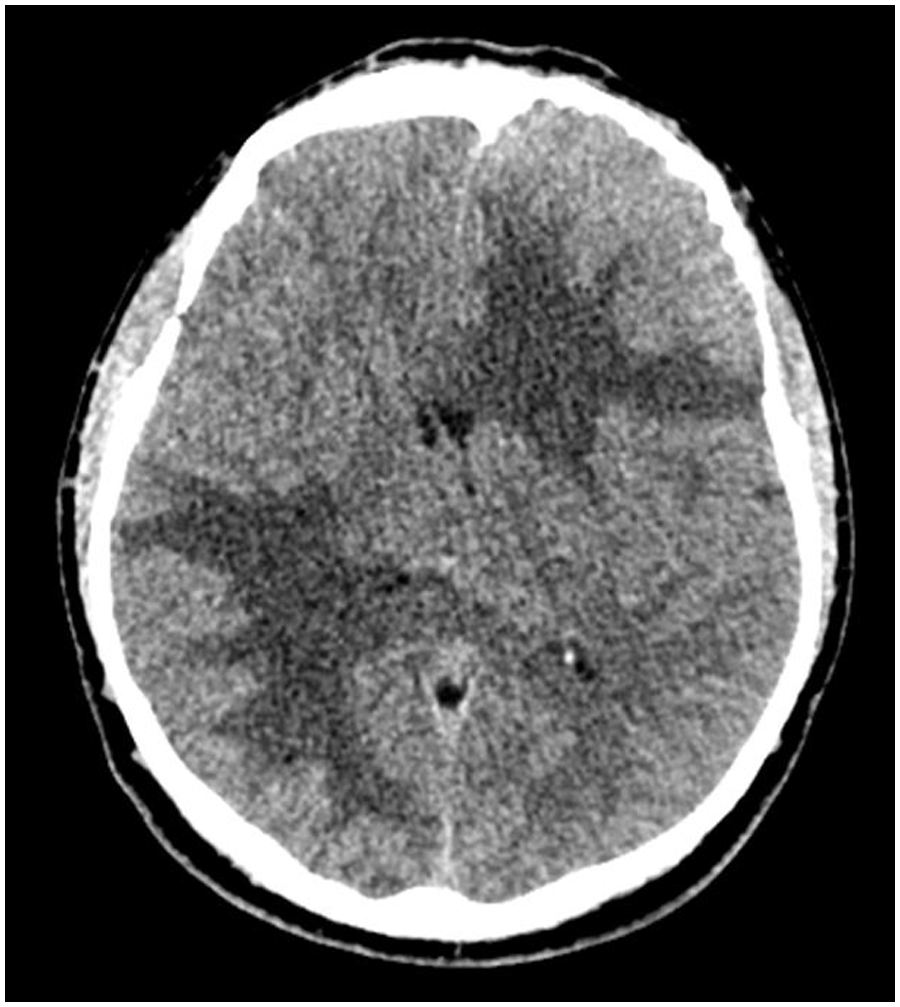
Non-injected cerebral CT showing an iso to hyper dense, extra-axial left frontal and right parieto-occipital processes.
The magnetic resonance imaging (MRI) that was made confirmed the meningeal starting point of these masses. T2-weighted sequence in coronal section and axial FLAIR showed a hyposignal meningeal masses. Axial sequence diffusion in b1000 revealed no restriction of diffusion. Gadolinium-injection demonstrated a strong and homogeneous enhancement (Figure 2).

(a) Axial FLAIR magnetic resonance imaging showing expansive bifocal lesions, isointense on FLAIR with mass effect and vasogenic type edema (arrow); (b) Iso intense signal on T2; (c) Axial gadolinium-enhanced image; and (d) Axial sequence diffusion in b1000 revealing no restriction of diffusion.
Surgical exploration was indicated. Intra-operative frozen section analysis of the tumor biopsy showed a very inflammatory process which led to the diagnosis of lymphoplasmacyte-rich meningioma. Thus, a total surgical resection of one mass was performed. Macroscopically, the lesion was firm; greyish-white and lobulated (Figure 3a). Microscopically, it was mostly constituted of large macrophages with eosinophilic or foamy cytoplasm and large vesicular nuclei. Neovascularization with a dense peri-vascular lymphocytic infiltrate was also noted. Many images of emperipolesis were observed, that were highly suggestive of a Rosai-Dorfman disease (Figure 3). The diagnosis was confirmed by the immunohistochemical profile of the histiocytes: S-100 protein+, CD68+, CD163+, Langerin−, and CD1a−. Immunostaining for BRAFV600E was negative (Figure 4).

(a) Macroscopic appearance of the mass; (b) Histologically, the lesion is constituted of large histiocytes having an eosinophilic and foamy cytoplasm (Hematoxylin-Eosin x20); (c) Many images of emperipolesis have been observed (arrow, Hematoxylin-Eosin x40); and (d) Note an inflammatory infiltrate rich in plasma-cells (Hematoxylin-Eosin x40).

At immunohistochemistry, histiocytes express S-100 protein (a) and CD163 (b).
No other localization of RDD was found. After surgery, oral corticosteroids (Prednisone commencing at 1 mg/kg/day) were used. The patient presented an excellent response with a significant regression and no increase in residual mass for 18 months.
Discussion and Conclusion
World Health Organization (WHO) defines Rosai-Dorfman disease as a clonal histiocytic proliferation characterized by large S100 positive histiocytes with variable emperipolesis. Recent molecular studies revealed recurrent mutations involving genes in the MAPK/ERK pathway suggesting a pathogenesis similar to Langerhans cell histiocytosis and Erdheim-Chester disease. 1
It predominantly affected children and young adult with a mean age of presentation of 20.6 years and with a slight male predominance.
Extranodal involvement is documented in 43% of the patients. 2 The CNS can be involved in less than 5% of the Rosai-Dorfman disease. In 90% of these cases the leptomeninges are affected.3,4
Clinically, the symptoms depend on the location such as cerebral convexity, parasagittal region, cranial base, suprasellar region, cavernous sinus, and petroclival region. They include cephalgia, seizure, or cranial nerve deficit.3,4
The typical radiological findings of intracranial RDD show dural-based extra-axial and well-circumscribed masses mimicking meningioma. Intracranial RDD CT typically presents a homogeneous hyperdense or isodense masses with possible perilesional edema, but MRI is currently the optimal diagnostic modality for evaluating lesions. On T2-weighted images, the lesions usually appear as isointense masses with intralesional hypointense foci. A lower T2 signal and the lack of hypervascularity on angiographic studies may help to differentiate it from meningioma.5,6
The definitive diagnosis of Rosai-Dorfman disease relies on a pathologic examination and immunohistochemical staining. 7
Microscopically, Rosai-Dorfman disease is presented as a multinodular mass. It is composed of a mixed inflammatory infiltrate including large pale histiocytes, numerous lymphocytes, plasma cells, and variable fibrosis. Emperipolesis with histiocytic engulfment of intact lymphocytes, plasma cells, neutrophils, and occasionally eosinophils is typical, but may be inconspicuous on Hematoxylin-Eosin staining. However, emperipolesis is not pathognomonic for Rosai-Dorfman disease and may occasionally be encountered in other neoplastic or non-neoplastic histiocytes and even in astrocytes. 1
At immunohistochemical study, RDD histiocytes are positive for CD163, CD68, and S100 antigens. RDD is CD1a and Langerin negative which distinguishes it from Langerhans cell histiocytosis. 1
Intra-operative pathological diagnosis can be a challenge. In our case, the technical artifacts of the frozen section and the density of the inflammatory infiltrate masked the typical lesions and pushed us to suggest the diagnosis of lymphoplasmacyte-rich variant of meningioma. This variant is a rare subtype of meningioma. It is characterized by an extensive chronic inflammatory infiltrate often overshadowing the inconspicuous meningothelial component. Neuroimaging features vary widely with frequent peri-tumoral edema and occasional multifocality or diffuse carpet-like meningeal involvement resembling pachymeningitis. 8 However, at immunohistochemical level, there are no similarities between the 2 entities. Meningothelial tumor cells express the Epithelial membrane antigen (EMA), Somatostatin receptor 2a (SSTR 2a), and Progesterone receptor (PR).
In addition to meningioma, the differential diagnosis of RDD is broad and can include IgG4-related disease, other types of histiocytosis such as Langerhans cell histiocytosis, lymphoproliferative disorders, and infectious disease. 9
Rosai-Dorfman disease of the CNS is preferably managed by surgical resection. Adjuvant therapies are taken into account when neurological symptoms persist or lesions appear around vital structures. There are different therapies including fractionated radiotherapy, stereotactic radiotherapy, corticosteroids, and chemotherapy, but more investigations need to be performed to find the optimal management of the disease. 10
Finally, the aim of our paper is to draw attention to this rare entity in young adult with multifocal masses mimicking meningioma. Meningeal lesion with predominant inflammatory changes should point to histiocytosis. Therefore, immunohistochemical staining for EMA, S100 and CD1a should be performed to ruling out the differential diagnosis.
The authors believe that the treatment strategy based in surgical resection and corticosteroids seems to be an effective therapy.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting OF interests:
THE author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All authors have read and approved final manuscript. HE wrote the manuscript; FT and ZIH participate in the development of radiological information; MH and MYO provided clinical and surgical information; FZ approved the diagnosis; AO, MJ, and NC supervised the work.