
Abstract
Background:
Tyrosine kinase inhibitors (TKIs) represent the standard of care for chronic myeloid leukemia (CML). Although cytopenias are frequent, severe and persistent bone marrow aplasia is exceedingly rare, with limited evidence to guide management in non-transplant candidates.
Case presentation:
A 35-year-old man with chronic-phase CML developed persistent bone marrow aplasia after sequential treatment with imatinib and dasatinib. Despite dose reductions and supportive measures, profound pancytopenia persisted, leading to treatment cessation. In the absence of a matched donor and response (BCR::ABL1: 25% IS), ponatinib was initiated at 45 mg daily and later reduced to 15 mg. Remarkably, hematologic recovery occurred, with normalization of peripheral blood counts, without recurrence of aplasia, and the patient achieved a sustained complete cytogenetic response within 15 months (BCR::ABL1 < 1% IS).
Discussion:
TKI-associated bone marrow aplasia has been reported with agents such as imatinib, dasatinib, and nilotinib. Proposed mechanisms include off-target inhibition of hematopoietic kinases (eg, c-KIT, PDGFR) and suppression of malignant clones preceding normal marrow recovery. Management typically involves discontinuation, transfusion support, immunosuppressive therapy, or transplantation. In this case, ponatinib proved effective and tolerable as salvage therapy after previous TKI-induced marrow aplasia.
Conclusion:
TKI-induced bone marrow aplasia is an uncommon but serious toxicity in CML. This case supports the potential role of ponatinib as an effective salvage option following severe TKI-related marrow toxicity, particularly in patients who are not candidates for allogeneic transplantation. Further clinical experience and larger studies are needed to better define optimal management strategies for this rare but serious complication.
Keywords
Introduction
Bone marrow aplasia and severe, persistent pancytopenia are rare but serious complications of Tyrosine Kinase Inhibitors (TKI) therapy, occasionally necessitating drug discontinuation and posing significant therapeutic challenges. Bone marrow aplasia can occur during treatment with TKIs, particularly in the first few weeks to months of therapy. 1 Previous studies have highlighted that this adverse event is attributable to the suppression of leukemia clones and inhibition of non-leukemic hematopoiesis. 2 Therefore, after a reduction in leukemic cell activity, normal stem cells may require time to regain their normal function. 2 This observation was supported by several prior studies, which indicate that the myelosuppressive effect is limited to the first few weeks to months of treatment. 3 Hence, with prolonged exposure to the TKI, the incidence of myelosuppression declines substantially. 3 Therefore, conventionally, when patients experience severe bone marrow aplasia, it is inevitable to discontinue the TKI agent. 3
Previous case reports have described comparable outcomes. Clinicians discontinued TKI therapy due to bone marrow aplasia.4,5 For instance, Kamijo et al 4 reported the discontinuation of imatinib, dasatinib, and ponatinib in a patient due to severe pancytopenia. Also, it was reported by Ramdial et al 5 that a patient who was started on multiple TKIs, including imatinib, dasatinib, and nilotinib, eventually developed bone marrow aplasia, leading to interruption of treatment. Despite the multiple treatment failures, the authors reported a successful outcome following bone marrow transplant, leading to an effective response.4,5 The optimal management of such cases is challenging and remains unclear; however, allogeneic stem cell transplantation is an established definitive treatment.
However, a significant challenge remains among patients who are ineligible for allogeneic stem cell transplantation. In this case report, we present the case of a young Indian male with chronic phase CML who developed profound and recurrent bone marrow aplasia with both imatinib and dasatinib, as he was not an eligible candidate for allogeneic stem cell transplant. However, upon initiation of ponatinib, he achieved hematologic and cytogenetic responses. To our knowledge, this is the first reported case of a successful outcome in a patient who developed bone marrow aplasia after prior TKIs (imatinib and dasatinib).
Case Presentation
A 35-year-old Indian male with no prior comorbidities was diagnosed with chronic-phase CML in November 2021 in India. He was started on imatinib therapy in India following diagnosis; however, detailed treatment records from this period were not available at the time of transfer of care in Qatar. He subsequently presented in December 2022 to the National Center for Cancer Care and Research (NCCCR) in Qatar with bilateral flank pain, shortness of breath, and splenomegaly. Laboratory tests revealed leukocytosis (leukocytes (WBC) 125 × 103/µL), hemoglobin (Hb) 14 g/dL, and platelet count 420 × 103/µL. Bone marrow (BM) aspirate and biopsy confirmed chronic-phase CML, showing BCR::ABL1 fusion in 97.5% of cells and an isolated Philadelphia chromosome t(9;22)(q34;q11.2) on karyotyping (Figure 1).

(A) CML at diagnosis; Bone marrow aspirate (50×) showing marked granulocytic hyperplasia, shift to left with no increase in blasts. BM biopsy (20×) showing marked hypercellularity (100%) with marked granulocytic hyperplasia (B).
Imatinib 400 mg daily was reinitiated in December 2022. However, shortly after treatment, the patient developed severe, recurrent pancytopenia requiring frequent red blood cell (RBC) and platelet transfusions, along with granulocyte colony-stimulating factor (G-CSF) support. Despite dose reductions, cytopenias persisted, prompting discontinuation of imatinib in October 2023. The patient’s complete blood count (CBC) recovered after discontinuation of imatinib, and dasatinib was initiated in October 2023. Within weeks, the patient again experienced profound pancytopenia (WBC 3.4 × 103/μL, Hb 6.7 g/dL, platelets 48 × 103/μL, RBC 1.8 × 106/μL, ANC 1.1 × 103/μL), accompanied by reticulocytopenia (absolute reticulocyte count 33.2 × 103/μL). Molecular response remained suboptimal, raising concern for drug-induced marrow suppression.
A repeat BM evaluation in March 2024 revealed marked hypocellularity (<5%), severely depressed trilineage hematopoiesis, and near-complete absence of megakaryocytes consistent with acquired amegakaryocytic thrombocytopenia. (Figure 2) BCR::ABL1 transcript level remained elevated at 25% (International Scale (IS)). No marrow fibrosis, dysplastic features suggestive of myelodysplasia, or cytogenetic evolution beyond the Philadelphia chromosome were identified. Clinical evaluation and laboratory investigations did not support autoimmune or infectious etiologies, making TKI-induced bone marrow aplasia the most likely diagnosis.
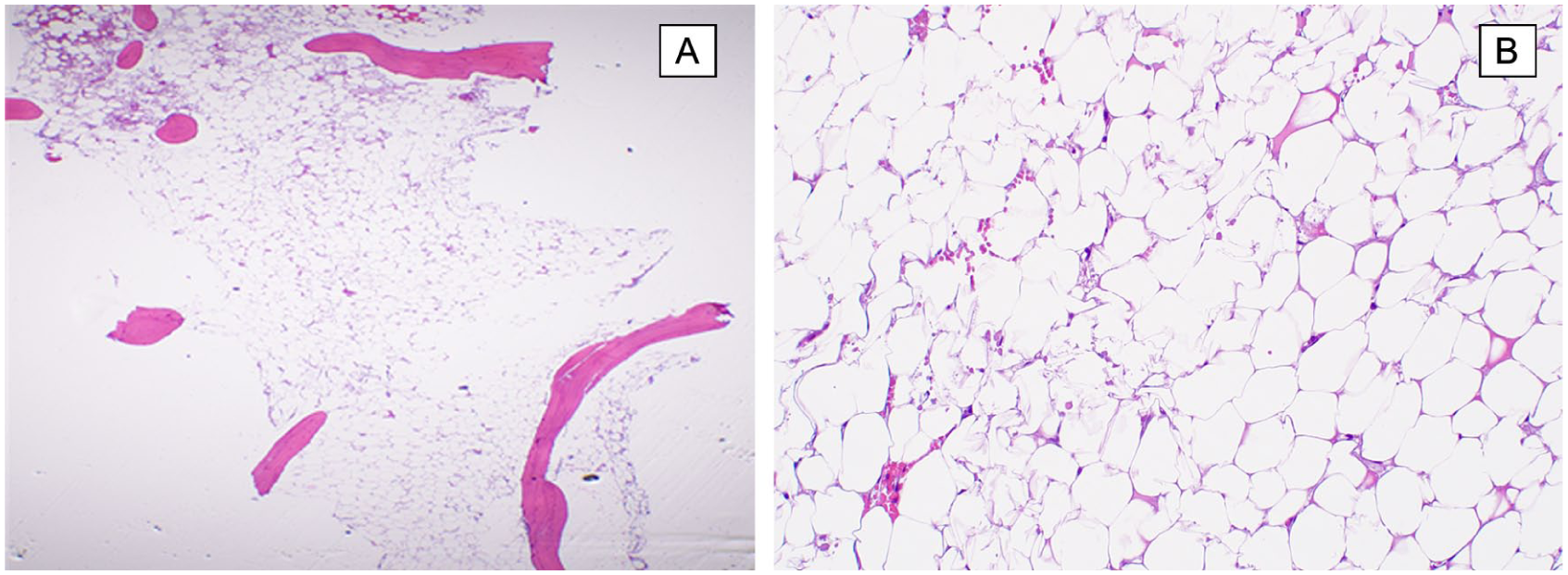
Bone marrow core biopsy (H&E) post treatment with imatinib and dasatinib showing severe marrow aplasia with markedly depressed trilineage hematopoiesis (<5% cellularity) (10×) (A) and 100× (B).
Given the failure of 2 prior TKIs and the unavailability of a matched donor for allogeneic stem cell transplantation, therapeutic options were limited. Ponatinib was initiated in June 2024 at 45 mg daily and was subsequently reduced to 15 mg after 2 months for tolerability. Unlike previous TKIs, ponatinib was well tolerated, with no recurrence of cytopenias. Improvement in peripheral blood counts was observed within the first months after initiation, followed by progressive normalization of hematologic parameters. Over a 15-month follow-up, the patient achieved sustained hematologic recovery, with stabilization of hemoglobin, leukocyte, and platelet counts. The BCR::ABL1 (p210) transcript levels declined to 0.9% IS, consistent with a molecular response corresponding to a complete cytogenetic response according to the European Leukemia Net (ELN) molecular surrogate criteria (Figure 3).

BCR::ABL1 transcript levels during sequential TKI therapy showing initial treatment failure with imatinib and dasatinib complicated by bone marrow aplasia, followed by successful molecular response with ponatinib salvage therapy.
Discussion
TKI-induced bone marrow aplasia is a rare complication, yet it is a serious adverse outcome seen among CML patients. Therefore, these adverse events pose a significant challenge for clinicians in managing CML and determining next steps for its control. The mechanism underlying this remains unclear; however, 2 main mechanisms have been proposed in the literature. First, off-target inhibition of kinases critical for normal hematopoiesis, particularly c-KIT and PDGFR, can impair the survival and proliferation of residual normal progenitor cells.4,6,7 Second, highly potent TKIs may eradicate malignant CML stem and progenitor clones so rapidly that remaining healthy progenitors are insufficient to sustain marrow function, leading to severe aplasia.4,6,8
Studies discussed the occurrence of bone marrow aplasia post TKI therapy among patients with chronic CML, highlighting the scarcity of data in relation to this adverse event. The challenge in this patient population arises from a lack of guidance on the appropriate management of CML patients with TKI-induced marrow aplasia. Several reports were published with various initiatives of optimal management. Starting with a case published by Kamijo et al, 4 describing a case of a 58-year-old CML patient who was initially treated with imatinib, followed by dasatinib, and then lastly ponatinib. Yet the patient consistently developed pancytopenia. Kamijo et al 4 reported that the patient was managed through haploidentical allogeneic hematopoietic stem cell transplantation, which successfully achieved a significant molecular response.
Similarly, Kassar et al 8 reported the occurrence of severe marrow hypocellularity consistent with an aplastic process in 2 patients after therapy with dasatinib. The authors reported unsuccessful outcomes following dose reduction and growth factor use in 1 patient. Ultimately, marrow aplasia was resolved by allogeneic stem cell transplant in one of the patients. 8 In 2021, Kassar et al. published a case of a 34-year-old female who developed profound marrow failure with marked hypocellularity post nilotinib therapy after treatment failure with imatinib. The authors reported the discontinuation of nilotinib and the use of immunosuppressive therapy. However, the patient passed away secondary to sepsis. 9
On the other hand, a 63-year-old female patient who developed similar adverse outcomes after imatinib therapy was indicated to stop the medication and was offered transfusion and supportive care, as reported by Lokeshwar et al. 10 Two published case reports by Lokeshwar et al and Khan et al reported that patients, unfortunately, died after developing bone marrow aplasia, where both authors reported the use of imatinib as first line TKI treatment.11,12 Further details on previous experiences with management of bone marrow aplasia are presented in Table 1.
Reported Cases of TKI-Induced Bone Marrow Aplasia in Patients with Chronic Myeloid Leukemia.

In our case, both imatinib and dasatinib induced severe and prolonged pancytopenia despite supportive measures. Ponatinib, a third-generation TKI with broader kinase inhibition and efficacy against resistant clones, was effective in this setting. The lack of alternative TKIs, persistent molecular disease, and ineligibility for allogeneic transplantation informed the decision to use ponatinib. However, ponatinib is associated with vascular risks; careful dose optimization and close monitoring allowed safe administration in this patient. The dose was reduced from 45 to 15 mg daily due to tolerability concerns; no recurrence of cytopenias or serious adverse events was observed. There is no clear evidence on the probability of marrow aplasia compared to the others. However, in our case report, the patient achieved a molecular response and hematologic recovery, supporting the proposed role of ponatinib as a rescue therapy when marrow toxicity limits other TKIs. While transplant remains a curative option, ponatinib may offer a viable bridge or alternative in patients without a suitable donor. As reported previously by Küçükyurt et al, 17 ponatinib was an optimal, less-toxic agent as a bridge to allogeneic stem cell transplantation in a CML patient in blast phase. Therefore, further research may be required to examine the impact of ponatinib in patients ineligible for transplantation.
Among the 10 reported cases, patients ranged in age from 35 to 73 years, with a clear male predominance (7 out of 10 patients with specified gender). All cases were in the chronic phase of CML. Prior treatment histories varied: some patients had received older cytoreductive therapies, such as hydroxyurea, busulfan, interferon-α, or low-dose cytarabine, whereas others had received TKIs as first-line therapy. None of the previously mentioned studies reported that ponatinib was effective post-TKI-induced marrow aplasia. On the contrary, some reports described further deterioration in hematologic parameters and overall clinical condition after exposure. While ponatinib use has been described in isolated cases of TKI-induced bone marrow aplasia, sustained hematologic recovery without the need for allogeneic transplantation and without recurrence of aplasia remains exceptionally uncommon.
The novelty of our case lies not in the first use of ponatinib, but in the sustained hematologic recovery, and continued tolerance of ponatinib without recurrence of bone marrow aplasia, thereby contributing additional clinical evidence supporting its potential role in carefully selected non-transplant candidates. This report describes a single patient’s experience, limiting the generalizability of these findings. Larger studies and additional clinical experience are needed to define the role of ponatinib in the management of TKI-induced bone marrow aplasia.
Conclusion
Bone marrow aplasia is an uncommon but potentially life-threatening adverse effect of TKI therapy in CML. The favorable hematologic recovery and molecular response observed highlight the potential of ponatinib as an alternative therapeutic option in cases of TKI-induced marrow toxicity, particularly when allogenic transplantation is not feasible. Further studies are needed to elucidate the underlying mechanisms and to guide optimal management of this rare but serious complication.
Footnotes
Acknowledgements
Not applicable.
Ethical Considerations
The case report was approved by the Hamad Medical Corporation’s Medical Research Center under the Number (MRC-04-25-1018). Written informed consent was obtained from the patient.
Author Contribution
R.G. (Writing, review and editing); L.S. (Writing, review and editing); R.Y.T. (Writing, review and editing); D.S. (Writing, review and editing); H.E. (Writing, review and editing); M.B. (Writing, review and editing); A.A. (Writing, review and editing); H.E. (Writing, review and editing).All authors read and approved the final version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Open Access funding provided by the Qatar National Library.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that support the findings of this study are not publicly available due to privacy reasons, but are available on reasonable request from the corresponding author.