
Abstract
Acquired von Willebrand syndrome (AVWS) is a rare clinical entity presenting with heterogeneous hemorrhagic manifestations, although some subsets of patients with AVWS may be asymptomatic until they are exposed to major trauma, an invasive procedure, or surgery. We herein report one such case in a 73-year-old male patient with nephrotic syndrome with a prolonged active partial thromboplastin time. We initially did not deal with this distinct abnormal clotting profile seriously, but persistent bleeding after a retroperitoneoscopic-assisted renal biopsy that allowed us to ascribe his nephrotic syndrome to membranous nephropathy fortuitously led to the discovery of concurrent AVWS. We feel that an accurate and prompt diagnosis as well as awareness of the disease remain a challenge for physicians and therefore strongly recommend the further accumulation of experiences similar to our own in a prospective manner. This report underscores the pitfalls associated with determining the bleeding risk, including an insufficient assessment and improper weighting of an abnormal clotting profile prior to the invasive procedure. Several management concerns that emerged in the current case are also discussed.
Keywords
Introduction
Acquired von Willebrand syndrome (AVWS) results from qualitative defects and/or the quantitative deficiency of von Willebrand factor (VWF), which plays a hemostatic role at the site of vascular injury. 1 Not surprisingly, it mimics the clinical and laboratory features of hereditary von Willebrand disease (VWD), presenting with heterogeneous hemorrhagic manifestations, with mucocutaneous bleeding being the most common.1–3 Of note, some subsets of patients with AVWS may be asymptomatic until they are exposed to major trauma, invasive procedures, or surgery. 4
We herein report one such case in a male patient complicated by nephrotic syndrome. Several management concerns that emerged in this case are also discussed.
Case Report
A 73-year-old man was admitted to our hospital complaining of progressive swelling of his lower extremities. Two months prior to this admission, the patient had recognized the leg manifestation when he was found to have a reduced serum albumin (Alb) level of 0.9 g/dL and 3+ proteinuria by his general practitioner despite having no apparent history of renal disease. Subsequently, the symptoms gradually worsened, and he was thus referred for a further workup. Other medical histories included bronchiectasis and permanent nonvalvular atrial fibrillation, which had been treated with clarithromycin and aspirin, respectively, for 7 years. There was no personal or family history of hereditary hemorrhagic disease.
At the time of admission (clinical day 0), the patient was alert with a temperature of 36.3°C and a blood pressure of 112/88 mm Hg, and he had gained approximately 5 kg in the previous 8 weeks, bringing his weight to 54.2 kg. He did not have pain in the lower extremities or dyspnea or tachypnea. Renal sonography showed that the right and left kidney long axis dimensions were 11.2 and 10.4 mm, respectively, with normal renal cortex echogenicity. A full blood count demonstrated a white blood cell count of 9900/µL (reference range: 3900-9800/µL) with normal differentials, a hemoglobin (Hb) level of 9.1 g/dL (reference range: 13.5-17.6 g/dL), and a platelet count of 44.0 × 104/µL (reference range: 13.0-36.9 × 104/µL), whereas a peripheral blood smear was unremarkable, and no atypical plasma cells were noted. Coagulation screening showed an active partial thromboplastin time (APTT) of 78.3 seconds (reference range: 23.1-36.3 seconds, control value: 29.9) with a prothrombin time-international normalized ratio (PT-INR) of 1.17 (reference range: 0.9-1.2). Further laboratory analyses revealed the following results: blood urea nitrogen, 32 mg/dL (reference range: 8-20 mg/dL); creatinine (Cr), 1.18 mg/dL (reference range: 0.63-1.03 mg/dL); total protein, 5.1 g/dL (reference range: 6.9-8.4 g/dL), Alb, 0.6 g/dL (reference range: 3.9-5.1 g/dL); total cholesterol, 181 mg/dL (reference range: 127-258 mg/dL); triglyceride, 63 mg/dL (reference range: 40-185 mg/dL); C-reactive protein, 4.82 mg/dL (reference range: 0.0-0.14 mg/dL); C3, 93 mg/dL (reference range: 86-160 mg/dL); C4, 23 mg/dL (reference range: 17-45 mg/dL), anti-double-stranded deoxyribonucleic acid antibodies, <5 IU/mL (reference range: <12 IU/mL); and fibrinogen/fibrin degradation product, 12.5 μg/mL (reference range: 0-5 μg/mL). Tests for hepatitis B virus surface antigens and antibodies to the hepatitis C were negative. Immunofixation electrophoresis of the serum and urine did not show any abnormalities. The patient’s urine contained 7.7 g of protein in a 24-hour specimen with a creatinine clearance of 31.6 mL/min. A diagnosis of nephrotic syndrome accompanied by a prolonged APTT was made; however, we did not deal with this distinct abnormal clotting profile seriously at this point.
Aspirin was discontinued on clinical day 13 and a retroperitoneoscopic-assisted renal biopsy, which allows for favorable hemostatic control under direct visualization because of its minimally invasive nature,5,6 was performed 9 days later (clinical day 22) as described with some modification. 5 The procedure was performed under general anesthesia with endotracheal intubation placing the Foley catheter in the bladder (to monitor the urine output) and a nasogastric tube. The first incision was performed along the elongation of the 12th rib at its intersection with the mid-axillary line, and minimal blunt dissection through the retroperitoneal fat was done. The peritoneum was then pushed forward to expose the lower pole of the left kidney. A second 5-mm trocar was placed on the paraspinal region at the level of the costovertebral angle. Through this second trocar, an 18-gauge gun-mounted semiautomatic biopsy needle was used to take multiple renal cortical tissue specimens. We applied pressure on the biopsy bed with a cotton tip dissector aiming for hemostasis, but protracted bleeding was encountered at the surgical site, leading to an estimated total blood loss during the procedure of 80 mL. The ports were finally removed under direct visualization after hemostasis was deemed satisfactory with a topical oxidized cellulose sheet, and the skin was closed with an absorbable suture in the usual standard technique using a Penrose drain.
However, the oozing of the wound persisted, and a laboratory analysis the next day revealed that the Hb level had decreased to 6.1 g/dL, which obliged us to perform empirical transfusions of fresh-frozen plasma as well as packed red blood cells. We therefore conducted several surveys to assess the concurrent coagulopathy. Due to the confirmation of a convex downward curve by the APTT cross-mixing test, which indicates the presence of a factor deficiency, 7 and a low factor VIII coagulant activity (FVIII:C) of 3.9% (reference range: 80%-140%), the patient was given a full-length recombinant factor VIII (50 IU/kg of rurioctocog alfa pegol) intravenously in accordance with the recommendation of a hematologist on clinical day 32. Around the same time, the sustained oozing gradually ceased; however, FVIII:C remained at 5.5% even after the supplementation of factor VIII. A reduced VWF ristocetin cofactor activity (VWF:RCo; <10%, reference range: 50%-150%) was later confirmed, and the multimeric analysis revealed the loss of large- to small-molecular-weight VWF multimers (Figure 1). A subsequent mixing experiment showed that the decline in the VWF:RCo of pooled normal plasma from 99% at baseline to 28% (2-hour incubation at 37°C with a ratio of the patient’s plasma to normal plasma of 1:1), suggesting the presence of inhibitors to VWF. Our patient was therefore regarded as having AVWS concurrently. Other evaluations, including abdominal ultrasound, thoracicoabdominal computed tomography, echocardiogram, and endoscopic analyses of the gastrointestinal tract, provided unremarkable findings. Finally, his nephrotic syndrome was ascribed to idiopathic membranous nephropathy based on the results of a systemic workup and renal pathologic assessment (Figure 2).
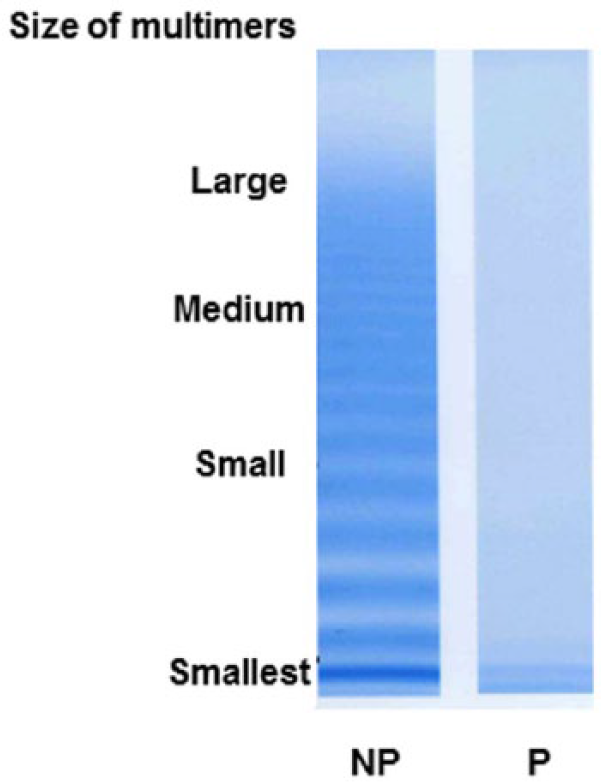
An electrophoretic analysis of von Willebrand factor (VWF) multimers. The patient’s plasma (P) multimer pattern is unusual and characterized by the presence of only a few bands around the corresponding region of smallest VWF protomer (bottom) without the discontinuous multimer pattern proper of pooled normal plasma (NP) VWF.
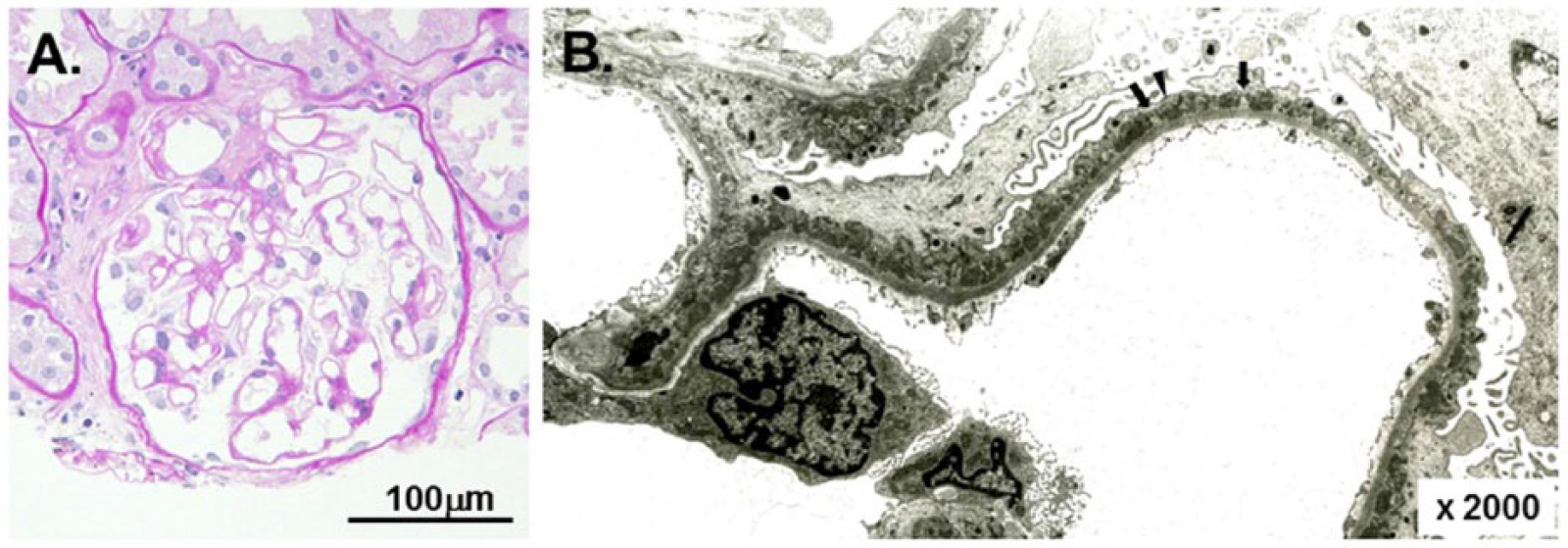
The renal biopsy findings. A light micrograph (A) shows the glomerulus with minor structural abnormalities (periodic acid-Schiff staining). The capillaries are patent, and the basement membranes are delicate. An electron micrograph of a portion of the glomerulus (B) demonstrates scattered segmental subepithelial electron-dense deposits (arrowhead) with distinct but sporadic intervening spikes (arrows), compatible with membranous nephropathy stage I to II. The scale or scale bar is indicated in each panel.
Oral prednisolone (PSL) in an alternate day fashion (mean daily dose of 15 mg) was commenced on clinical day 40. Neither the resumption of aspirin nor the commencement of warfarinization was done because the patient refused to receive preventive medication for thromboembolism secondary to atrial fibrillation despite a thorough discussion. While no abnormalities were noted in the patient’s serum-free T4 level (1.3 ng/dL, reference range: 0.84-1.44 ng/dL), despite a slightly decreased free T3 level of 1.77 pg/mL (reference range: 2.11-3.51 pg/mL) on clinical day 53, a laboratory analysis performed on clinical day 93 still showed a prolonged APTT of 52.3 seconds (control value: 29.9) with marginally improved FVIII:C at 10.5% and VWF:RCo at 21%. A normalized APTT of 34.3 seconds (control value: 29.9), FVIII:C of 95.8%, and VWF:RCo of 56% were finally confirmed on clinical day 232. The dosage of PSL was titrated thereafter at mean daily doses that ranged from 5 to 15 mg, depending on the context.
The patient was doing well for the next 4 years until he abruptly developed right hemiparesis with a maximum deficit, which was attributed to left temporo-occipital lobe ischemic stroke. At this point, he still had nephrotic syndrome with a serum Alb level of 2.5 g/dL; however, a normal coagulation profile, including an APTT of 32.7 seconds (control value: 29.9), FVIII:C of 130.2%, and VWF:RCo of 158%, was noted although the clotting parameters had not been monitored on a regular basis. The patient was therefore treated with intravenous heparin and then switched to warfarin, delivering a PT-INR of 1.5 to 2.5. His neurologic symptoms were relieved without any bleeding complications, and only slight muscle weakness remained in his upper limb 15 days after the onset of stroke.
Eventually, the patient was lost to follow-up as his care was transferred to the regional geriatric hospital. During the observation period, his serum Cr levels slowly but steadily increased to around 1.6 mg/dL, whereas heavy proteinuria of approximately 6 to 7 g/gCr persisted, and his nephrotic syndrome never reached even partial remission.
Discussion
This report illustrates a rare case in which persistent bleeding after a retroperitoneoscopic-assisted renal biopsy performed as a diagnostic investigation of nephrotic syndrome fortuitously led to the discovery of concurrent AVWS. We feel that an accurate and prompt diagnosis as well as awareness of the disease remains a challenge for physicians, despite the accumulation of studies concerning the nature of AVWS.1–4,8,9 Postoperative bleeding because of a hemorrhagic defect after undergoing routine surgical procedures is a well-recognized complication; however, the possibility of an isolated coagulation factor defect may not necessarily be suspected in a timely manner, especially in elderly patients. 10 Furthermore, a prolonged APTT may be occasionally ignored in ordinary clinical practice, as occurred preoperatively in the present case. Although we have no specific way to predict hemorrhagic events, 11 our experience underscores the pitfalls associated with determining the bleeding risk, including an insufficient assessment and improper weighting of an abnormal clotting profile prior to various kinds of invasive procedures.
AVWS is an uncommon heterogeneous bleeding disturbance characterized by variedly low plasma levels of VWF and factor VIII, occurring principally in individuals without a personal or family history of hemorrhagic diathesis.1,2 Various chronic illnesses, including neoplasms, congenital or acquired cardiovascular defects, immunologic disturbances, and hypothyroidism, have been shown to be associated with this disease.2,3 Lymphoproliferative and myeloproliferative disorders may be the most common underlying pathologies, whereas miscellaneous conditions responsible for AVWS include Epstein-Barr viral infection, uremia, and therapeutic use of several agents such as ciprofloxacin, griceofulvin, and valproic acid.2–4 Reduced VWF synthesis, inactivation or accelerated clearance of VWF by paraproteins, adsorption of VWF high-molecular-weight multimers, and increased proteolytic degradation of VWF have received focus as pathogenic bases for the disease.2,4 Compared with the acquired form of hemophilia A, which is exclusively due to auto-antibodies against factor VIII, 7 the disease process of AVWS is more heterogeneous, and none of the proposed mechanisms appears to be specific to the different underlying conditions.2,4 Since it was first described as a separate entity in a patient with systemic lupus erythematosus in 1968, 8 the cumulative number of publications on AVWS has been slowly growing, and at present, about 600 cases have been identified in the international literature. 9 The incidence of the disease remains to be clarified, as the current data are largely limited to retrospective studies, and the number of patients with AVWS may be underdiagnosed2,4,9; however, Kumar et al reported their experience showing that up to 4.5% of patients with a clinical and laboratory diagnosis of VWD may have the acquired form, giving an estimated AVWS incidence of as much as 0.04% in the general population. 12 A number of such patients are advanced in age, but all age groups have been affected—from 2 to 96 years of age with a median age of 62 years—with no distinct sex predominance.2,12,13
Distinguishing AVWS and VWD is important in terms of the differences in their therapeutic strategies. 14 A late onset of hemorrhagic events and no family history of coagulopathy should prompt suspicion of AVWS, although FVIII:C, VWF:RCo, the plasma levels of VWF antigen (VWF:Ag), and the VWF-collagen-binding activity (VWF:CBA) are occasionally diminished in patients with AVWS.3,4,9,13 Patients with a reduced ratio of VWF:RCo/VWF:Ag and/or VWF:CBA/VWF:Ag may have a selective loss or decrease in high-molecular-weight VWF multimers.3,9,14 Such quantitative abnormalities can be visually confirmed by a multimer analysis using electrophoretic separation and immunostaining.14,15 Mixing tests coupled with an assay of VWF:RCo can detect inhibitors of VWF among some subsets of patients with AVWS; however, this method does not allow us to detect relevant nonneutralizing antibodies, and an enzyme-linked immunosorbent assay detecting a broader spectrum of VWF-binding antibodies may provide beneficial information, where available.1,9,14
However, despite the benefits of these analyses, none are diagnostically specific, and some of these tests are even technically demanding, preventing their routine implementation in ordinary clinical settings. 9 Treating the underlying pathologies when they can be identified has been a plausible therapeutic option for AVWS. 14 Such management strategies include surgery, chemotherapy, radiotherapy, and the cessation of the offending agents, and patients with hypothyroidism may respond well to thyroxine therapy.4,9,14 Other options include the administration of glucocorticoids, desmopressin, VWF-containing concentrates, plasmapheresis, and high-dose immunoglobulin given intravenously.2–4,9,14 However, no consensus regarding the optimum therapeutic regimens has been established, with present approaches depending on each patient’s conditions. Furthermore, treating the underlying disorders is not always successful, and even reaching complete or partial remission may not necessarily be associated with symptomatic or laboratory improvements in patients with the disease.4,14
In the current case, the patient’s clinical manifestations, a careful medical record review, a thorough clinical interview, and systemic evaluations hampered our identification of the pathology listed among the disease states associated with AVWS.2–4,9,13,16 Whether or not there is an causal link between AVWS and idiopathic membranous nephropathy in the present patient remains unclear. Although our patient did not achieve remission of his nephrotic syndrome even after the commencement of steroid treatment, which is not very different from previous experiences, 17 our patient seemed to benefit from the procedure in terms of the favorable improvement in his abnormal coagulation profiles, implying that immune system disturbance played a role, at least in part, as a pathogenic basis of the AVWS in the present patient. Consequently, the clinical scenario of the present patient characterized by such a favorable outcome may not be surprising. However, the significance of the present case should be evaluated carefully, given his concurrent nephrotic syndrome due to membranous nephropathy and atrial fibrillation that might have necessitated management decisions on coagulation prophylaxis for thromboembolic events.18–20
Patients with nephrotic syndrome have been shown to be at an increased risk of venous and/or arterial thrombosis, including cerebral infarction.18,19,21,22 The pathogenic basis for an accelerated clotting activity in this population is likely to depend on a net shift in the hemostatic equilibrium toward a hypercoagulable state, resulting from abnormal processing of prothrombotic and antithrombotic proteins. 23 However, pharmacologic coagulation prophylaxis, which may have been empirically practiced among patients with nephrotic syndrome due to varied glomerulopathies,19,23–25 has not been accepted as the standard of care, and a selective or individualized approach still seems justified in the field of nephrology. 18 Given the clinical features of ischemic stroke characterized by the sudden onset of maximum neurologic deficit, 26 we believe that atrial fibrillation-mediated cardioembolism contributed in a major way to the disease onset in the present patient, although the concurrent nephrotic syndrome might also have played a role additively or synergistically. Our failure to prescribe any agents aimed at prophylactic anticoagulation before the onset of the stroke may ultimately invite criticism; however, no consensus regarding such a management among patients with AVWS, who may be relatively protected against various kinds of thromboembolism, has yet been established. This precluded us from evaluating the patient’s risks of stroke and bleeding that occurred as a result of anticoagulation using standard scoring systems, as is practiced in the general population.20,27 We were thereby obliged to give priority to the patient’s request. Given that systemic studies on this topic are lacking, we strongly recommend the prospective accumulation of more experiences similar to our own. We believe that evaluating similar cases will help clarify the overall management strategies for thromboembolic prophylaxis among patients with acquired bleeding disorders as well as clarify the nature of AVWS.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by a Grant-in-Aid for Research on Advanced Chronic Kidney Disease, Practical Research Project for Renal Diseases from the Japan Agency for Medical Research and Development (AMED).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
TS and TA drafted the manuscript. TK and TY made contributions to the acquisition of the clinical data. EK and DN provided a detailed review of the contents and structure of the manuscript, resulting in significant changes to the original document. All of the authors have read and approved the final manuscript.
Disclosures and Ethics
As a requirement for publication, the authors have provided the publisher with signed confirmation of their compliance with legal and ethical obligations including, but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) the protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this manuscript is unique and not under consideration for publication or published in any other journals and that they have permission from the rights holders to reproduce any copyrighted material. The external blind peer reviewers report no conflicts of interest.