
Abstract
In our article, we asked whether Sox2, a transcription factor important in brain development and disease, is involved in gene regulation through its action on long-range interactions between promoters and distant enhancers. Our findings highlight that Sox2 shapes a genome-wide network of promoter-enhancer interactions, acting by direct binding to these elements. Sox2 loss affects the three-dimensional (3D) genome and decreases the activity of a subset of genes involved in Sox2-bound interactions. At least one of such downregulated genes, Socs3, is critical for long-term neural stem cell maintenance. These results point to the possibility of identifying a transcriptional network downstream to Sox2, and involved in neural stem cell maintenance. In addition, interacting Sox2-bound enhancers are often connected to genes which are relevant, in man, to neurodevelopmental disease; this may facilitate the detection of functionally relevant mutations in regulatory elements in man, contributing to neural disease.
Identifying genes involved in neurodevelopmental defects is a major ongoing endeavor. Previously, catalogs of human brain enhancers and of polymorphisms within them have been produced and used to identify potential connections to schizophrenia and autism.1–4 On the other hand, genes functionally connected to these enhancers have just begun to be defined. 5 In our article, 6 we started to identify functional connections between genes and enhancers in the mouse genome, with a view to provide a basis for analysis of neural disease-relevant genes in humans.
Sox2 Function in the Transcriptional Regulation of Neural Cell Identity and Disease
We focused on the roles of the transcription factor Sox2 in regulating the genome-wide interactions between genes and enhancers, thus shaping the global three-dimensional (3D) interactome. 6 Sox2 is necessary for the maintenance of both embryonic stem cells (ESCs) 7 and neural stem cells (NSCs), both in vivo and in vitro. 8 In man, Sox2 mutation results in hippocampal defects, eye defects, intellectual disability, and other neural defects.
The Role of Noncoding Elements in Regulating Development and Disease
Extensive effort has identified thousands of active enhancers in human and mouse neural cells using DNase sensitivity or chromatin immunoprecipitation of genomic sequences, bound by p300 or modified histones like H3K27ac, coupled with sequencing (ChIP-seq). These foundational atlases of neural enhancer identities have been a major contribution to the field of neural development and disease, but these studies did not reveal the specific gene promoter(s) that are directly regulated by each enhancer. Indeed, interactions between enhancers and promoters cannot be understood using the aforementioned approaches, as these methods can only delineate the linear location of active enhancers along the length of a chromosome. However, the mammalian genome is organized in three dimensions through a complex looping arrangement that is relevant to transcriptional regulation.1,9 Inside the nucleus, chromosomes are extensively folded into chromatin loops that occupy distinct 3D territories. This highly organized 3D chromatin organization provides a topological basis for many genome functions, including transcription, by bringing distal regulatory elements and their cognate genes into close spatial proximity. 9 Chromatin organization and enhancer-promoter interactions are highly dynamic during lineage specification.10,11 Long-range interactions connect genes (or gene promoters) to other promoters (promoter-promoter interactions), or to distant regions (promoter-enhancer interactions) carrying enhancer marks, often skipping other intervening genes.
The Use of Chromatin Interaction Assays in Wild-Type and Sox2-Deleted Brain-Derived NSC to Determine Sox2-Associated Interactions
At the genome level, these interactions (interactome) have been extensively characterized by techniques such as HiC and Chromatin Interaction Analysis by Paired-End-Tag sequencing (ChIA-PET), which rely on the detection, by high-throughput sequencing, of proximity ligated DNA fragments within fragmented fixed chromatin.2, 9 ,11,12 These studies provided extensive catalogs of potential enhancers (and polymorphism within them), connected in various tissues to specific genes. At the genome-wide level, the regulatory functions of the detected enhancers and interactions have been so far addressed by few previous studies.2,5,12
We adopted RNApolII ChIA-PET (as well as an improved version of it, “in situ ChIA-PET”) to capture genome-wide chromatin interactions between genes and enhancers that are associated with RNA polymerase II (RNAPII) and, concomitantly, to identify previously unrecognized enhancer elements in the mouse genome. We thus analyzed mouse brain-derived wild-type and Sox2-mutant neonatal NSC grown in vitro (Figure 1A). In parallel, we identified the genome-wide localization of DNA-bound Sox2, together with regions carrying histone modifications indicative of enhancer activity (H3K27Ac; H3K4me1). Overall, Sox2-bound interactions are about 36% to 46% of the total interactions, pointing to a potential role of Sox2 in the generation or maintenance of the NSC interactome. DNA-bound Sox2 is highly enriched within “enhancer regions” genome-wide; in particular, the proportion of total Sox2-bound “enhancers” that are represented within DNA fragments (anchors) involved in promoter-enhancer interactions is much higher than that of non-Sox2-bound “enhancer regions” (Figure 1B), pointing to a potential role of Sox2 specifically at the level of these interactions.
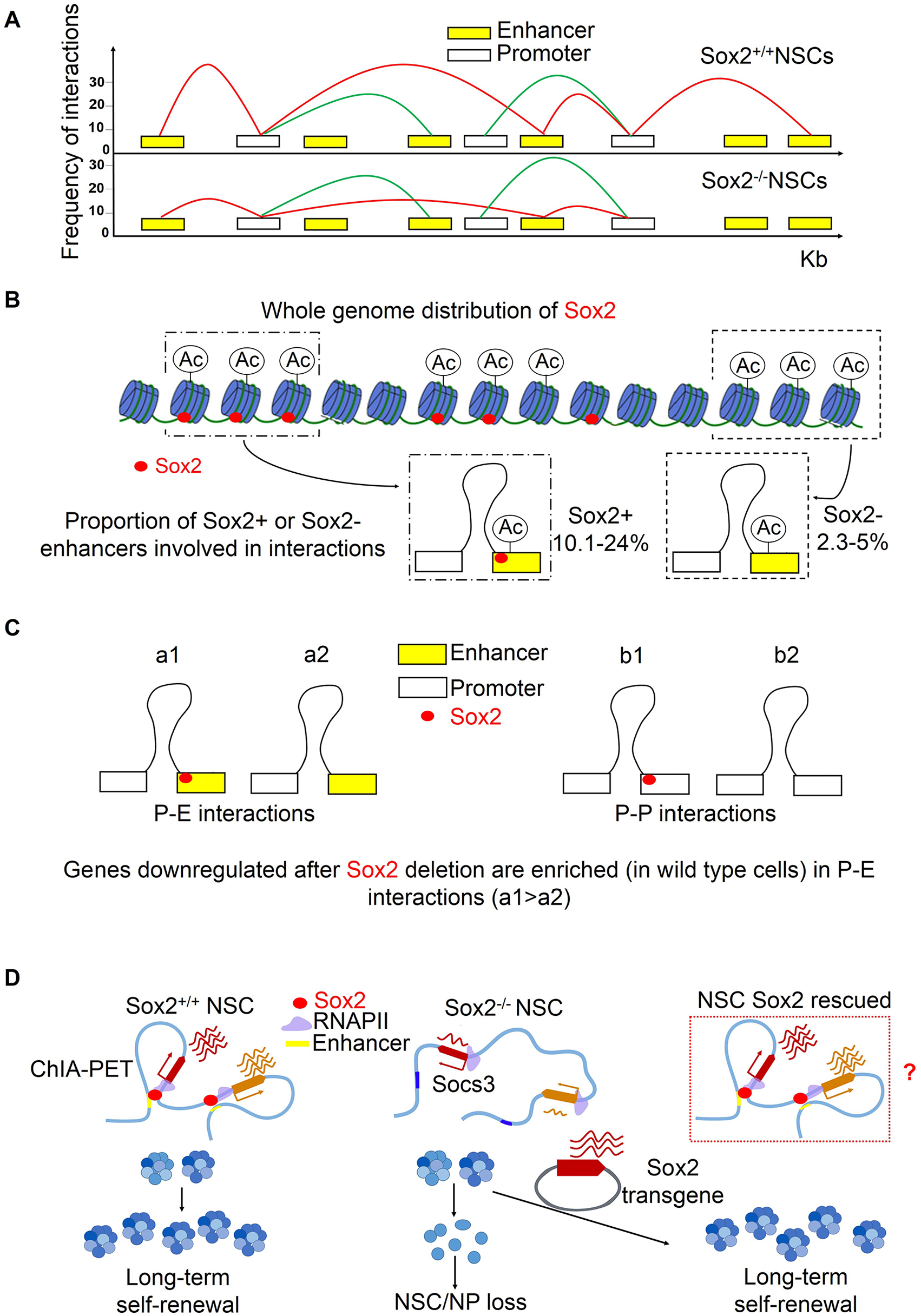
Sox2-dependent interactions: (A) promoter-enhancer and promoter-promoter interactions are depicted in wild-type (top) and Sox2-deleted (bottom) NSC. Red indicates interactions decreased in mutant cells. (B) Sox2 binding overlaps with H3K27Ac+ nucleosomes (“enhancers”). The proportion of Sox2+ enhancers in interactions is much higher than the proportion of Sox2– enhancers (see text). (C) The loss of Sox2 from enhancers involved in promoter-enhancer interactions is the major factor responsible for downregulation of the connected gene. (D) Is loss of long-range interactions in Sox2-deleted NSC rescued by re-addition of Sox2, together with long-term self-renewal?
Sox2 Deletion Profoundly Affects the Pattern of Interactions in Neonatal NSC
In neonatal cultures obtained from the brain of mice in which Sox2 had been deleted during embryogenesis, the interactions are, in general, greatly reduced: some are largely lost, whereas others are less importantly affected in frequency (Figure 1A). The strong decrease in the frequency of some of the interactions is indicative of a complete loss in a significant proportion of the cells.
The parallel study of the transcriptome indicates that Sox2-deleted cells show significantly decreased expression of some 700 genes. We looked for a correlation between the type of interaction identified for each downregulated gene, and its belonging to the category of genes that are downregulated in mutant NSC, versus nondownregulated genes. The “downregulated genes in mutant” category is quite significantly enriched in promoter-enhancer interactions, and even more in promoter-Sox2+ enhancer interactions (in wild-type cells), whereas the “nondownregulated” category shows anticorrelation with these characteristics (Figure 1C). These results indicate that the activities of a significant fraction of genes are dependent on their interaction with an enhancer, in particular an Sox2-bound enhancer. Note that Sox2 deletion causes significant alterations of the expression of several transcription factors, which might in turn contribute to the changes in the interactome; so, the observed correlation of gene downregulation (in mutant cells) with gene interactions with Sox2-bound enhancers (in wild type cells) emerges on top of a background of effects not directly dependent on Sox2.
A similar analysis shows that promoter-promoter interactions do not importantly affect the expression of genes in NSC, although their loss may correlate with minor decreases of expression in a subset of genes. Overall, our results stress the importance of promoter-enhancer interactions for transcriptional regulation and indicate that the predominant functional effect of Sox2 on gene transcription is exerted by its binding to distal enhancers connected to regulated genes, rather than to promoters.
It is presently unclear whether Sox2 acts directly to bring about an interaction. It is possible that Sox2 acts by recruiting other transcription factors, generating complexes which form bridges connecting different DNA regions; additionally, Sox2 might interact with structural factors able to create/maintain loops. YY1, a noncell type-specific factor, promotes the formation of a tissue-specific interactome in neural cells by binding to specific sequences 13 : it is tempting to speculate that YY1 may collaborate with Sox2 in these processes. It will be interesting to identify the binding of additional transcription factors in the proximity of Sox2 bound to anchors.
A large proportion of the genes involved in interactions with enhancers maintain their activity even after Sox2 loss; moreover, alterations in chromatin epigenetic marks are not detected in Sox2-deleted cells. There are several possible explanations: (1) many genes have multiple interactions, and the more interactions a gene has, the greater its activity (on average). As not all of the interactions centered on a given gene are necessarily lost in Sox2-deleted cells, it is possible that the remaining interactions are sufficient to confer significantly high activity; (2) the maintenance of an “active gene” epigenetic configuration might contribute to preserve gene activity. This hypothesis implies that Sox2 might be required for the establishment of the active chromatin configuration, and of gene activity, that might then be maintained, upon later deletion of Sox2, by additional mechanisms (eg, binding of other transcription factors recruited by the initial Sox2 binding); (3) interactions detected by ChIA-PET might represent a “snapshot” of a process (chromatin connectivity) that may be dynamic; (some) interactions might be successively in an “off” or “on” state, in which case, loss of Sox2 might simply have altered (ie, decreased) the probability of persisting in, or entering into, the “on” state (resulting in the observed overall decrease of interactions).
Identification of Critical Genes for Sox2-Dependent NSC Maintenance
Why do Sox2-deleted NSC lose the ability to self-renew? We looked for genes that are likely directly regulated by Sox2 (ie, bound by Sox2 at the enhancer or promoter in NSC, or stimulated by Sox2 in transgenic zebrafish assays), and that lose a large part of their expression in Sox2-deleted cells. Among several genes sharing these characteristics, we identified Socs3 (suppressor of cytokine signaling), whose deletion is known to favor diversion of NSC into the glial differentiation pathway. 6 We then transduced Socs3, together with a green fluorescent protein (GFP) reporter gene, into Sox2-deleted NSC; this resulted in a progressive increase of the proportion of GFP+ cells, up to almost 100%, and the efficient long-term maintenance of the growth of these cells, well beyond the time when untransduced Sox2-deleted cells had all been lost from the culture. It is likely that additional genes will be identified by similar screens; these might either cooperate with Socs3, or might represent genes upstream to Socs3, and active, together with Sox2, on Socs3 regulation, thus defining a network of Sox2-dependent functional interactions.
The fact that Socs3 transduction rescues the maintenance of Sox2-deleted NSC raises the interesting question whether the altered long-range interactions pattern, and the correct gene expression levels, can be restored to normal in rescued Sox2-deleted NSC. Initial work indicates that Sox2 transduction is also able to rescue NSC self-renewal; thus, the loss of self-renewal is reversible in Sox2-deleted cells. ChIA-PET experiments will address the issue of restoration of the correct interactome in these cells (Figure 1D).
Human Neural Disease-Relevant Gene Regulatory Networks and Identification of Functional Neural Enhancers
Our data offer an exciting potential strategy to identify pathogenic noncoding variants affecting the function of their connected genes involved in neural diseases in man. Many of the brain enhancers identified in our studies show one or more of these characteristics: (1) they stimulate forebrain expression of a reporter gene in zebrafish; (2) they coincide with enhancers active in early forebrain development in mouse 14 ; and (3) they are highly significantly connected to genes whose human homologs are responsible, when mutated, for intellectual disability, microcephaly, vision disorders, Alzheimer and Parkinson diseases, and so on (Figure 2). The large catalog of the presently identified mouse distal “enhancers” may allow to look for mutations and polymorphisms in the corresponding human enhancers (if conserved) to facilitate the search for genetic variation responsible for brain inherited disease and for predisposition to schizophrenia and autism spectrum disorders.

Long-range Sox2-bound interactions and human-inherited neurodevelopmental disease: (A) functional enhancers connected to mouse homologs of human neural disease genes may provide a clue to the identification of mutation/polymorphism in conserved human enhancers, responsible for disease. (B) Mouse homologs of genes involved in neurodevelopmental disease in man are significantly enriched in Sox2-bound promoter-enhancer interactions. Different types of microcephaly and intellectual disability are indicated. The proportion of Sox2-bound (red) to total genes considered (blue) is indicated (see Bertolini et al, 6 Table S6). (C) Screenshots of interactions involving the PTEN-induced kinase 1 (PINK1) and Presenilin-2 (Psen2) genes, involved in Parkinson’s and Alzheimer’s disease, respectively.
Perspectives for More In-depth Studies of the Functional Interactome
While HiC and ChIA-PET provide an extensive catalog of (potentially) all the interactions, they have some limitations. These techniques detect multiple gene promoters brought together through chromatin interactions from the collection of large number of cells. Two different models can account for these aggregated chromatin interactions: specific, pairwise promoter-enhancer interactions, or multivalent, in parallel chromatin interactions between different promoters and multiple regulatory modules. That is, the enhancer activity could be target-specific, combinatorial, or both (Figure 3). Moreover, some of the region-specific interactions are identified by large numbers of sequencing reads, whereas other interactions are rare; the latter ones are likely present only in a small proportion of the cells in the population. An assessment of functional roles of the interaction would ideally require the knowledge of which interactions are present simultaneously on each gene region in a given cell. Such an analysis has been made recently possible (in Drosophila cells) by a new technique (ChIA-Drop). 15 In ChIA-Drop, individual chromatin complexes are partitioned into droplets with single gel bead containing unique DNA-barcoded primers, such that all the chromatin fragments within each droplet have the same barcodes and can be distinguished following linear amplification. The amplified barcoded DNAs are then pooled for high-throughput sequencing. Sequencing reads having identical barcodes are grouped as multivalent and represent combinatorial chromatin interactions between each enhancers with all corresponding genes targets (Figure 3) which identifies fragments present within single “drops” containing individual chromatin fragments from single cells. 15 If this technique can be applied to mouse or human cells, it will allow additional understanding of the function of the interactome. This will be particularly interesting when analyzing the simultaneous interactions of the enhancer component, revealing whether interaction of a given enhancer is, at any specific time, limited to a single gene, or extended to multiple different genes.

Two models for aggregated chromatin interactions.
Footnotes
Acknowledgements
The authors thank the members of their laboratories and their collaborators (in particular S. Ottolenghi, G. Pavesi, P. Bovolenta) for discussion and support.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Nicolis laboratory is supported by ERANET NEURON funding (ImprovVision grant, NEURON8-Full-815-091). M.P. is the recipient of a fellowship from the Italian Ministry of University and Research (MIUR) through the grant “Dipartimenti di Eccellenza-2017” to the University of Milano Bicocca, Department of Biotechnology and Biosciences. This publication was also partially supported by the 4DN (U54 DK107967), R01 GM127531-01A1 and ENCODE (UM1 HG009409) consortia from National Institute of Health, U.S.A. to YZ an CLW.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SKN and CLW wrote the article; MP and YZ (who had done fundamental experiments in the discussed research paper) discussed the article, and prepared the Figures.