
Abstract
The zinc finger protein ZPR1 deficiency causes neurodegeneration and results in a mild spinal muscular atrophy (SMA)-like disease in mice with reduced Zpr1 gene dosage. Mutation of the survival motor neuron 1 (SMN1) gene causes SMA. Spinal muscular atrophy is characterized by the degeneration of the spinal cord motor neurons caused by chronic low levels of SMN protein. ZPR1 interacts with SMN and is required for nuclear accumulation of SMN. Patients with SMA express reduced levels of ZPR1. Reduced Zpr1 gene dosage increases neurodegeneration and severity of SMA disease in mice. Mechanisms underlying ZPR1-dependent neurodegeneration are largely unknown. We report that neurodegeneration caused by ZPR1 deficiency is mediated by the c-Jun NH2-terminal kinase (JNK) group of mitogen-activated protein kinases (MAPK). ZPR1-dependent neuron degeneration is mediated by central nervous system (CNS)-specific isoform JNK3. ZPR1 deficiency activates the MAPK signaling cascade, MLK3 → MKK7 → JNK3, which phosphorylates c-Jun and activates caspase-mediated neuron degeneration. Neurons from Jnk3-null mice show resistance to ZPR1-dependent neurodegeneration. Pharmacologic inhibition of JNK reduces degeneration of ZPR1-deficient neurons. These data show that ZPR1-dependent neurodegeneration is mediated by the JNK signaling pathway and suggest that ZPR1 downregulation in SMA may contribute to JNK-mediated neurodegeneration associated with SMA pathogenesis.
Introduction
The ZPR1 gene is evolutionary conserved and is essential for cell viability in eukaryotes.1-4 ZPR1 is ubiquitously expressed in mammalian cells and mediates receptor tyrosine kinases (RTKs) signaling by binding to the cytoplasmic domain of epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptor (PDGFR) that are highly conserved among family of RTKs. 5 ZPR1 binds to inactive EGFR and PDGFR in quiescent mammalian cells, and treatment with mitogen or serum results in translocation of ZPR1 from the cytoplasm to the nucleus and supports cell growth and proliferation.2,5 ZPR1 interacts with eukaryotic translation elongation factor 1A (eEF1A) and ZPR1-eEF1A protein complexes are required for normal cell growth and proliferation. 1 ZPR1 also interacts with survival motor neuron (SMN) protein. ZPR1 is required for nuclear accumulation of SMN in sub-nuclear bodies, including gems and Cajal bodies (CBs).3,6 The severity of spinal muscular atrophy (SMA) disease correlates negatively with the number of SMNs containing sub-nuclear bodies. 7 Interaction of ZPR1 with SMN is disrupted in cells derived from patients with SMA. Spinal muscular atrophy is the leading cause of infant mortality caused by homozygous deletion or mutation of the SMN1 (telomeric) gene.8,9 A second copy SMN2 (centromeric), which is similar to SMN1, undergoes alternative splicing and produces ~10% of full-length SMN protein and ~90% truncated product lacking exon 7 (SMNΔ7).10,11 Chronic low levels of SMN result in degeneration of spinal cord motor neurons followed by muscle atrophy leading to respiratory failure and death in SMA.12,13
The expression of ZPR1 is downregulated in patients with SMA, and the low levels of ZPR1 cause defects in accumulation of SMN in sub-nuclear bodies.6,14,15 ZPR1 deficiency causes defects in mRNA biogenesis, including splicing and transcription.3,6,16 The reduced expression of ZPR1 causes neurodegeneration, increases respiratory distress, and contributes to severity of disease in SMA mouse models.15,17,18 However, the molecular mechanism of neurodegeneration caused by ZPR1 deficiency is largely unknown. In this study, we examined the effect of ZPR1 knockdown on cultured primary cerebellar granule neurons (CGNs) and the spinal cord motor neurons and investigated the role of the c-Jun NH2-terminal kinase (JNK) group of mitogen-activated protein kinase (MAPK) kinases in mediating ZPR1-dependent neurodegeneration. We demonstrate that the JNK signaling pathway mediates neurodegeneration caused by ZPR1 downregulation and inhibition of JNK activity slows/prevents degeneration of neurons.
Materials and Methods
Mice
The wild-type (FVB/N) mice were purchased from the Jackson Laboratory. The Jnk3–/– knockout mice 19 on C57BL/6 background were backcrossed for 10 generations to wild-type FVB/N mice to create Jnk3–/– knockout mice on FVB background. 20 Wild-type and Jnk3–/– knockout mice breeding was established to generate 7-day-old pups. Genotyping of mice for the Jnk3 gene was performed by polymerase chain reaction (PCR) using tail DNA. 20 Mice were euthanized to collect tissues, cerebellum, and the spinal cords for culture of CGNs and spinal motor neurons, respectively, and biochemical analysis. All experiments and procedures were approved and performed according to the guidelines and policies set by the Institutional Biosafety Committee. All animals were housed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC), Texas Tech University Health Sciences Center El Paso, El Paso, Texas. Animals were treated humanely, and euthanasia was performed using methods approved by the American Veterinary Medical Association.
MAPK array analysis
The phospho-MAPK antibody array was used for the identification of specific isoform of JNK and processed according to manufacturer’s protocol (R&D Systems Inc.). Protein extracts were prepared from cultured CGNs as described below. Array images were analyzed using densitometry and ImageJ software. Relative signal intensities normalized to β-actin or glyceraldehyde 3-phosphate dehydrogenase (GAPDH; mean ± SEM) were represented as bar graphs. 20 The likely molecular targets were tested and confirmed by biochemical methods using mouse cultured primary neurons. To reduce biological variation, protein extracts of cultured CGN from 3 mice were pooled and examined by phospho-MAPK antibody arrays.
Primary neuron culture and RNAi
Primary CGNs were isolated from the cerebellum of 7-day-old wild-type and Jnk3–/– mice and cultured in vitro for 6 days in 35-mm dish or 8-well chamber microscope slides, coated with poly-
IB analysis
Protein extracts were prepared from cultured neurons using Triton lysis buffer. 6 To reduce biological variation 2 to 4 control or treated neuron samples were pooled to prepare protein extracts. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to the PVDF membrane (Millipore). IB analysis was performed using primary antibodies (1:1000) followed by horseradish peroxidase (HRP)-conjugated donkey anti-mouse or rabbit IgG (1:5000) secondary antibodies. The following primary antibodies were used for probing blots: ZPR1 (clone #LG-C61), 18 SMN (Clone 8; Transduction Lab), cleaved Caspase-3 (Asp175) #9661 (Cell Signaling), alpha-tubulin (Sigma), Phospho-MKK7 (Ser271/Thr275) #4171, Phospho-MLK3 (Thr277/Ser281) #2811, Phospho-SEK1/mitogen-activated protein kinase kinase 4 (MKK4) (Ser257/Thr261) #9156, Phospho-SAPK/JNK (Thr183/Tyr185) (81E11) #4668, Phospho-c-Jun (Ser63) #9261, and c-Jun (60A8) #9165 (Cell Signaling). Chemiluminescence and quantitation of IBs were performed using ImageQuant LAS4000. The relative amounts of proteins (mean ± SEM) normalized to tubulin are presented as bar graphs.
IF analysis
Spinal cord neurons and CGNs were isolated, purified from the spinal cords and cerebellum of 7-day-old wild-type and Jnk3–/– mice, respectively, and cultured in vitro for 6 days in 8-well chambers coated with poly-
Statistical analysis
The quantitation of data is presented as mean ± SEM. Statistical analysis performed using either one-way analysis of variance (ANOVA) or Student’s t-test (unpaired, 2-tailed) with GraphPad Prism (version 5.0d). The value P = .05 or less was considered significant. In all experiments, “n” represents the number of times the experiment was performed. A minimum of n = 3 number of times each experiment was performed, unless otherwise specified in an experiment.
Results
ZPR1 deficiency causes caspase-mediated neuron degeneration
To unravel the molecular mechanisms of neurodegeneration caused by ZPR1 deficiency, we examined the effect of ZPR1 deficiency on the activation of MAPKs and components of cell death signaling pathways. The primary CGNs were isolated from the cerebellum of 7-day-old mice and cultured in vitro for 6 days. 21 Cultured primary neurons were mock-transfected or transfected with control siRNA (Scramble) or Zpr1-specific siRNA (siZPR1). Primary neurons were harvested 72 h post transfection and examined by IB and IF analyses. The quantitative and statistical analyses of IBs show Zpr1-specific siRNA results in about 75% decrease and ZPR1 levels reduced to 24.91% ± 3.271%, n = 3, P = .0001 in treated neurons (siZPR1) compared with control (untreated) or treated with scrambled siRNA (Scramble; Figure 1A and B). ZPR1 knockdown in neurons causes small but significant decrease in SMN levels 19.18% ± 5.481% (P = .0217), suggesting that reduced SMN levels may have minor contribution to neuron degeneration caused by ZPR1 deficiency (Figure 1C).

ZPR1 deficiency causes Caspase 3-mediated degeneration of cultured cerebellar granule neurons. (A) Primary cerebellar granule neurons from 7-day-old mice were cultured for 6 days, and mock-transfected (control), transfected with scrambled siRNA (Scramble) and ZPR1-specific siRNA (siZPR1). Effect of ZPR1 knockdown on the levels of ZPR1, SMN, activated* (cleaved) caspase 3, and α-tubulin (72 h post transfection) was examined by immunoblot analysis using specific antibodies. Biological variation was reduced by pooling 2 to 4 control or siRNA-treated samples. (B and C) Quantitative and statistical analyses of protein levels after ZPR1 knockdown. (B) ZPR1 knockdown with siRNA results in low levels of ZPR1 (24.91% ± 3.271%, n = 3, P = .0001) in neurons that show ~75% reduction in levels of ZPR1. (C) ZPR1 knockdown results in small but significant decrease in SMN levels 19.18% ± 5.481% (P = .0217). (D) The Zpr1 gene suppression causes axonal defects in primary cerebellar granule neurons from 7-day-old mice. Primary neurons mock-transfected (Control) and transfected with scrambled siRNA (Scramble) and ZPR1-specific siRNA (siZPR1) for 72 h, fixed with 4% PFA, and stained with antibodies to neuron-specific class III β-tubulin (red) and ZPR1 (green). (E) Neurons stained for neuron-specific class III β-tubulin (red) and SMN (green). (F) Neurons stained for neuron-specific class III β-tubulin (red) and Cytochrome C (Cyto C) (green). (G) Neurons stained for neuron-specific class III β-tubulin (red) and activated* (cleaved) Caspase 3 (green). Nuclei were stained with DAPI (blue). Stained neurons were examined by confocal microscopy. Scale bar is 20 µm. DAPI indicates 4′,6-diamidino-2-phenylindole; JNK, c-Jun NH2-terminal kinase; PFA, paraformaldehyde; SMN, survival motor neuron.
Immunofluorescence analysis shows that knockdown of ZPR1 (siZPR1) causes severe axonal degeneration compared with control and scramble-treated neurons (Figure 1D). ZPR1 deficiency causes mislocalization of SMN and accumulation of SMN in the cytoplasm as indicated by arrowheads in the siZPR1 panel (Figure 1E). These results are consistent with our previous findings and show that ZPR1 is required for nuclear accumulation of SMN.3,6 It is possible that mislocalization of SMN caused by ZPR1 deficiency may also contribute to neuron degeneration. Next, we examined the markers of apoptotic neuron death in ZPR1-deficient neurons. Knockdown of ZPR1 results in a marked increase in the staining of Cytochrome C (Cyto C) compared with control and scramble siRNA-treated neuron (Figure 1F). Immunoblot analysis shows increase in the levels of activated (cleaved) Caspase 3* (Figure 1A), which is supported by increased staining of activated Caspase 3* in ZPR1-deficient degenerating neurons (Figure 1G). These data suggest that ZPR1 deficiency causes activation of Caspase 3-mediated apoptotic pathway, which leads to neuron degeneration and death.
ZPR1 deficiency causes activation of the JNK signaling pathway in neurons
To determine whether ZPR1 deficiency causes JNK activation, we first examined the effect of ZPR1 knockdown on the phosphorylation of c-Jun, which is an established downstream target of JNK. The phosphorylation of c-Jun was not detected in neurons treated with scrambled siRNA (control). In contrast, neurons treated with ZPR1-specific siRNA (siZPR1) show robust increase in phosphorylation of c-Jun and its accumulation in the nucleus (Figure 2A). To determine whether the phosphorylation of c-Jun is caused by JNK activation, we examined primary neurons lacking ZPR1 by staining with phospho-specific antibodies against activated JNK. ZPR1 deficiency results in phosphorylation (activation) and nuclear accumulation of JNK (Figure 2B). Together, these data show that ZPR1 deficiency results in activation of JNK that causes phosphorylation of c-Jun and triggers Cyto C release and Caspase 3 activation leading to apoptotic degeneration of neurons.

ZPR1 deficiency causes activation of the JNK signaling pathway. (A) Primary cerebellar granule neurons from 7-day-old mice were cultured for 6 days, and mock-transfected (Control), transfected with scrambled siRNA (Scramble) and ZPR1-specific siRNA (siZPR1) for 72 h, fixed with 4% PFA and stained with antibodies to neuron-specific class III β-tubulin (red) and phospho-c-Jun (p-c-Jun) (green). (B) Neurons stained for neuron-specific class III β-tubulin (red) and phospho-JNK (p-JNK) (green). Nuclei were stained with DAPI (blue). Stained neurons were examined by confocal microscopy. Scale bar is 20 µm. DAPI indicates 4′,6-diamidino-2-phenylindole; JNK, c-Jun NH2-terminal kinase; PFA, paraformaldehyde.
To test whether ZPR1 deficiency also causes activation of JNK in the spinal cord motor neurons and mediates degeneration, we examined primary cultured spinal cord neurons from 7-day-old mice. We have shown previously that cultured primary spinal cord neurons retain characteristics of motor neurons and stain positive for motor neuron markers, including ChAT and homeobox Hb9 (Hlxb9 or Hb9).15,20,22 Mock (Control) and siRNA-treated (siZPR1) neurons stained with antibodies against ZPR1 and neuron-specific β-tubulin show that knockdown of ZPR1 causes degeneration, including axonal retraction and ballooning of spinal cord neurons (Figure 3A). Analysis of neurons stained with phospho-JNK (p-JNK) shows low levels of staining in control neurons compared with robust staining of p-JNK in the soma and the nucleus of degenerating ZPR1-deficient neurons (siZPR1; Figure 3B). The presence of p-JNK in ZPR1-deficient neurons suggests activation of JNK. Further, examination of c-Jun, a downstream target of activated JNK, shows that c-Jun is heavily phosphorylated (p-c-Jun) and accumulated in the nucleus of degenerating ZPR1-deficient neurons (siZPR1) compared with control neurons (Figure 3C). These data show that the ZPR1 deficiency causes activation of the JNK cascade in the spinal cord motor neurons. These data suggest that the low levels of ZPR1 reported in patients with SMA6,14 may contribute to JNK pathway-mediated neurodegeneration in SMA. 20

ZPR1 deficiency causes activation of the JNK in the spinal cord motor neurons. (A) Primary spinal cord neurons from 7-day-old mice were cultured for 12 to 14 days, and mock-transfected (Control), transfected with scrambled siRNA (Scramble) and ZPR1-specific siRNA (siZPR1) for 72 h, fixed with 4% PFA, and stained with antibodies to neuron-specific class III β-tubulin (red) and ZPR1 (green). ZPR1 knockdown causes axonal degeneration, including bending, retraction, and ballooning. (B) ZPR1 knockdown causes increase in the levels of phospho-JNK (p-JNK) in the soma and the nucleus of motor neurons treated with siZPR1. Neurons stained for neuron-specific class III β-tubulin (red) and p-JNK (green). (C) Phosphorylation of JNK in ZPR1-deficient neurons results in its activation, and p-JNK phosphorylates downstream target c-Jun. Motor neurons stained for β-tubulin (red) and phospho-c-Jun (p-c-Jun) (green). Nuclei were stained with DAPI (blue). Stained spinal cord motor neurons were examined by confocal microscopy. Arrowheads indicate axonal degeneration. Scale bar is 20 µm. DAPI indicates 4′,6-diamidino-2-phenylindole; JNK, c-Jun NH2-terminal kinase; PFA, paraformaldehyde.
The JNK group of kinases is activated by dual phosphorylation on threonine and tyrosine by an upstream MAPK kinase (MAPKK or MAP2K), which in turn is activated by an MAPKK kinase (MAPKKK or MAP3K) that mediates extracellular or intracellular stress responses. The specificity of activation of the JNK signaling cascades such as MAPKKK → MAPKK → JNK1/2/3 is achieved by scaffold JNK-interacting proteins (JIPs).24,25 To identify upstream MAPKKs that are known to activate JNK, 26 we examined activation of MKK4 and MKK7 using phospho-specific antibodies. Analysis of phospho-MKK4 (p-MKK4) shows that MKK4 is not activated in ZPR1-deficient neurons (Figure 4A). Notably, ZPR1 knockdown causes activation of MKK7 and shows the presence of p-MKK7 in ZPR1-deficient (siZPR1) compared with control or scramble-treated neurons (Figure 4B). Selective activation of MKK7 by ZPR1 deficiency shows specificity of ZPR1-mediated degeneration of neurons. The mixed lineage kinase 3 (MLK3) belongs to MAPKKK family and known to activate JNK in CGN and mediate stress-induced apoptosis. 27 The examination of activation of MLK3 using antibody against phospho-MLK3 (p-MLK3) shows activation of MLK3 in ZPR1-deficient neurons compared with scramble-treated neurons (Figure 4C). These data suggest that ZPR1 deficiency activates MAPK cascade MLK3 → MKK7 → JNK that mediates degeneration of cerebellar neurons.

ZPR1 deficiency causes activation of the mitogen-activated protein (MAP) kinases upstream of JNK kinase. (A) Primary cerebellar granule neurons from 7-day-old mice were cultured for 6 days, and mock-transfected (control), transfected with scrambled siRNA (Scramble) and ZPR1-specific siRNA (siZPR1) for 72 h, fixed with 4% PFA and stained with antibodies to neuron-specific class III β-tubulin (red) and phospho-MAP kinase kinase 4 (p-MKK4) (green). (B) Neurons stained for neuron-specific class III β-tubulin (red) and phospho-MAP kinase kinase 7 (p-MKK7) (green). (C) Neurons stained for neuron-specific class III β-tubulin (red) and phospho-mixed lineage kinase 3 (p-MLK3) (green). Nuclei were stained with DAPI (blue). Stained neurons were examined by confocal microscopy. Scale bar is 20 µm. DAPI indicates 4′,6-diamidino-2-phenylindole; JNK, c-Jun NH2-terminal kinase; PFA, paraformaldehyde.
ZPR1-dependent neuron degeneration is mediated by JNK3
Immunoblot analysis of p-JNK and p-c-Jun shows that ZPR1 deficiency may activate different isoforms of JNK (Figure 5A). Mammals have three Jnk1, Jnk2, and Jnk3 genes that encode JNK isoforms. 24 However, which JNK isoform is required for neuron degeneration caused by the loss of ZPR1 is unclear. To identify the specific JNK isoform that mediates neurodegeneration, we used purified cultured neurons and performed phospho-MAPK array analysis. The MAPK array analysis shows small increase in activation of JNK1 and JNK2, and a marked increase (25-fold, n = 4, P = .0001) in the phosphorylation of JNK3, a neuron-specific isoform, 19 in ZPR1-deficient degenerating neurons (Figure 5B). These data show that the low levels of ZPR1 cause activation of JNK3 in cultured primary neurons. These data suggest that ZPR1-dependent neurodegeneration is mediated by the MLK3 → MKK7 → JNK3 signaling pathway.

Neuron-specific isoform JNK3 is required for neuron degeneration caused by ZPR1 deficiency. (A) Cultured mouse CGNs were transfected with scrambled siRNA (Control) or ZPR1-specific siRNA (siZPR1). Neurons were harvested 72 h post-transfection for analysis of protein extracts using immunoblot analysis. ZPR1 deficiency causes increase in phosphorylation of JNK isoforms (p-JNK2 and p-JNK1,3) and phospho-c-Jun (p-c-Jun). (B) ZPR1 deficiency causes activation of JNK3, a neuron-specific isoform, in cultured primary neurons. The levels of phosphorylation of JNK isoforms were examined by phospho-MAPK antibody array. The levels of phosphorylation of individual JNK isoforms, JNK1, JNK2, and JNK3, were quantitated by densitometry and relative intensities were calculated (mean ± SEM) to plot fold activation as a bar graph. Quantitative analysis shows marked increase (25-fold, n = 4, P = .0001) in JNK3 phosphorylation (activation) compared with JNK1 and JNK2 phosphorylation. (C) JNK3 is required for neuron degeneration caused by ZPR1 deficiency. JNK3 deficiency reduces neuron degeneration caused by low levels of ZPR1. Primary neurons (CGN) from wild-type (Jnk3+/+) and knockout (Jnk3–/–) 7-day-old mice were transfected with scrambled (Control) or ZPR1-specific siRNA (siZPR1) and cultured for 72 h, fixed with 4% PFA and stained with antibodies to β-tubulin (red) and phospho-JNK (green). ZPR1 knockdown results in activation (phosphorylation) of JNK (green) in siZPR1-treated Jnk3 (+/+) neurons compared with control Jnk3 (+/+) neurons (columns 1 and 2). Knockdown of ZPR1 in JNK3-deficient neurons from Jnk3 (–/–) knockout mice did not cause activation of JNK (green) and show reduced degeneration of neuron compared with Jnk3 (+/+) neurons (column 3). (D) Neurons from knockout Jnk3–/– mice were transfected with plasmid expressing GFP-JNK3 and with either scrambled (Control) or ZPR1-specific siRNA (siZPR1), cultured for 72 h and stained with antibodies to β-tubulin (red). JNK3 was examined by GFP fluorescence (green) using confocal microscopy. Nuclei stained with DAPI (blue). Scale bar is 20 µm. CGN indicates cerebellar granule neuron; DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; PFA, paraformaldehyde.
To test whether JNK3 is required for neurodegeneration, we examined the effect of ZPR1 deficiency on neurons lacking JNK3. Neurons from wild-type (Jnk3+/+) and JNK3 knockout (Jnk3–/–) mice were cultured and transfected with siRNA. The knockdown of ZPR1 results in activation of JNK (p-JNK) and degeneration of Jnk3+/+ neurons (Figure 5C, tubulin panel). Notably, p-JNK was not detected in Jnk3–/– neurons lacking ZPR1 and degeneration of Jnk3–/– ZPR1-deficient neurons was reduced compared with Jnk3+/+ ZPR1-deficient neurons (Figure 5C). These data show that JNK3-deficient neurons are resistant to ZPR1-dependent neurodegeneration suggesting that JNK3 deficiency reduces degeneration of neurons with the low levels of ZPR1. To further test whether JNK3 is required for degeneration of neurons, we complemented Jnk3–/– neurons with recombinant GFP-JNK3 23 using transient transfection and examined the effect of ZPR1 deficiency. Control experiments show that the GFP-JNK3 was distributed in the axons and soma of Jnk3–/– neurons. (Figure 5D). Interestingly, knockdown of ZPR1 (siZPR1) results in degeneration of Jnk3–/– neurons expressing recombinant GFP-JNK3 (Figure 5D). These data suggest that JNK3 is required for neurodegeneration caused by ZPR1 deficiency. Together, these data suggest that JNK3 mediates neurodegeneration caused by ZPR1 deficiency and JNK3 represents a potential therapeutic target to reduce/prevent neurodegeneration caused by the low levels of ZPR1 in patients with SMA.
Inhibition of JNK prevents ZPR1-dependent neuron degeneration
Genetic inactivation of the Jnk3 gene in neurons results in reduced degeneration of ZPR1-deficient neurons suggesting that pharmacologic JNK inhibition may provide neuroprotection (Figure 5). To test whether pharmacologic inhibition of JNK will reduce neuron degeneration, we examined the effect of extensively studied JNK inhibitor (SP600125). 28 Control experiments show that the treatment with SP600125 (1 µM) did not affect ZPR1 distribution or levels and integrity of neurons (Figure 6A, tubulin panel). Comparison of neurons lacking ZPR1 (siZPR1) treated with solvent (DMSO) and inhibitor (SP600125) shows that the JNK inhibition results in reduced degeneration of ZPR1-deficient neurons (Figure 6A). To test whether reduced degeneration of ZPR1-deficient neurons is a result of inhibition of JNK activity, we examined activation of c-Jun (p-c-Jun) in ZPR1-deficient neurons (siZPR1) treated with DMSO and SP600125. Neurons lacking ZPR1 (siZPR1) show marked increase in phosphorylation of c-Jun (p-c-Jun) compared with control neurons (Figure 6B). The treatment with SP600125 shows inhibition of JNK activity that is represented by decrease in p-c-Jun staining in neurons (SP600125) compared with ZPR1-deficient neurons treated with DMSO (Figure 6B, p-c-Jun). Treatment with SP600125 also shows reduced degeneration of ZPR1-deficient neurons, suggesting a correlation between JNK inhibition and protection of ZPR1-deficient neurons (Figure 6B). To measure the potential neuroprotective effect of genetic and pharmacologic JNK inhibition on ZPR1-deficient neurons, we performed quantitative analysis of neuron survival with and without JNK inhibition. Quantitative and statistical analyses show knockdown of ZPR1 levels by (73.68% ± 4.82%, P = .0001) results in degeneration (75.37% ± 2.50%, n = 4, P = .0001) (siZPR1) of neurons compared with control neurons and about 25% neurons survive (Figure 6C). Comparison of ZPR1-deficient neurons treated with DMSO and SP600125 shows that pharmacologic inhibition of JNK results in reduced degeneration (~31%) and increased (68.06% ± 2.53%, P = .0001) survival of neurons (Figure 6C). Furthermore, comparison of ZPR1-deficient neurons from Jnk3+/+ (Control) and Jnk3–/– mice shows that genetic inhibition of JNK results in reduced degeneration (~21%) and increased (79.06% ± 2.53%, P = .0001) survival of neurons (Figure 6C). Together, these data suggest that genetic and pharmacologic inhibition of JNK results in a marked and statistically significant reduction of neuron degeneration caused by ZPR1 deficiency.

Inhibition of JNK prevents ZPR1-dependent neuron degeneration. (A) The inhibition of JNK decreases degeneration of neurons expressing low levels of ZPR1. Primary neurons transfected with scrambled siRNA (Control) and ZPR1-specific siRNA (siZPR1) were treated with DMSO or SP600125 after 36 h (post-transfection). Neurons were incubated for additional 60 h, fixed, and stained with antibodies to tubulin (red) and phospho-JNK (p-JNK) (green). Nuclei stained with DAPI (blue). Stained neurons were examined by confocal microscopy. Scale bar is 20.0 µm. (B) Inhibition of JNK kinase activity results in decreased phosphorylation of c-Jun and reduced degeneration of ZPR1-deficient neurons. Primary neurons transfected with scrambled siRNA (Control) and siZPR1 were treated with DMSO or SP600125 after 36 h (post-transfection). Neurons were incubated for additional 60 h, fixed, and stained with antibodies to tubulin (red) and phospho-c-Jun (green). Nuclei were stained with DAPI (blue). Stained neurons were examined by confocal microscopy. Scale bar is 20.0 µm. (C) Quantification of neuron degeneration. Neurons stained with tubulin (red) and nuclei stained with DAPI used for quantitative analysis. Neurons with intact axons were counted in Control (transfected with scrambled), siZPR1 (treated with ZPR1-specific siRNA), siZPR1 + SP600125 (1 µM), and siZPR1 + Jnk3–/– (neurons lacking JNK3). Forty images, 10 each from 4 different experiments for control or treated were examined for neuron degeneration. The average (mean ± SD; n = 4) percent of surviving neurons stained with nuclear DAPI is plotted as a bar graph. The statistical significance (P-value) of neuroprotection data was determined by 1-way ANOVA and unpaired t-test (2-tailed). Knockdown of ZPR1 results in degeneration of neurons (75.37% ± 2.50%, P = .0001) (siZPR1). Pharmacologic inhibition of JNK with JNK inhibitor SP600125 treatment increases survival (68.06% ± 2.53%, P = .0001) of ZPR1-deficient neurons (siZPR1 + SP600125) compared with siZPR1-treated neurons (21.83% ± 2.37%). Genetic inhibition of JNK with knockout of the Jnk3 gene results in increase in survival (79.06% ± 2.53%, P = .0001) of ZPR1-deficient neurons (siZPR1 + Jnk3–/–) compared with siZPR1-treated neurons (21.83% ± 2.37%). ANOVA indicates 1-way analysis of variance; DAPI, 4′,6-diamidino-2-phenylindole; DMSO, dimethylsulfoxide; JNK, c-Jun NH2-terminal kinase; PFA, paraformaldehyde.
Discussion
The ZPR1 gene is evolutionary conserved in yeast and mammals. The human ZPR1 is located on Chr11q23.2 and encodes a multi-domain protein, including two Zn2+ fingers, a domain that connects two zinc fingers (A-domain) and a COOH-terminal domain (B-domain) that have unique features and involved in mediating protein and nucleic acid interactions.1,4,6,18 Gene knockout studies in yeast and mice have shown that the ZPR1 is essential for cell and embryonic viability, respectively.1-3 However, essential function of ZPR1 is unknown. ZPR1 may contribute to the normal function of SMN 29 because ZPR1 deficiency results in mislocalization of small nuclear spliceosomal proteins (snRNPs), a major target of SMN activity, and causes defects in pre-mRNA splicing and transcription.3,6,16 Recent studies have shown that ZPR1 may be a putative transcription factor that regulates light-regulated genes in plants and developmentally-regulated Hox genes in mammals.18,30 Patients with SMA express the low levels of ZPR1.6,14,15 Genetic studies with reduced Zpr1 gene dosage have demonstrated that the low levels of ZPR1 cause neurodegeneration 17 and increase the severity of disease and reduce the lifespan of mice with SMA. 15 The molecular mechanisms that may mediate neurodegeneration caused by the chronic low levels of SMN in SMA have been reported and summarized in a recent review. 31 However, the mechanism of neurodegeneration caused by the low levels of ZPR1 is unclear. In this study, we demonstrate that the JNK signaling pathway mediates the neurodegeneration caused by the low levels of ZPR1. A graphical summary and the mechanism of activation of the JNK signaling pathway in ZPR1-deficient neurons leading to neurodegeneration and the prevention of ZPR1-mediated neurodegeneration using genetic and pharmacologic inhibition of JNK are shown in Figure 7.
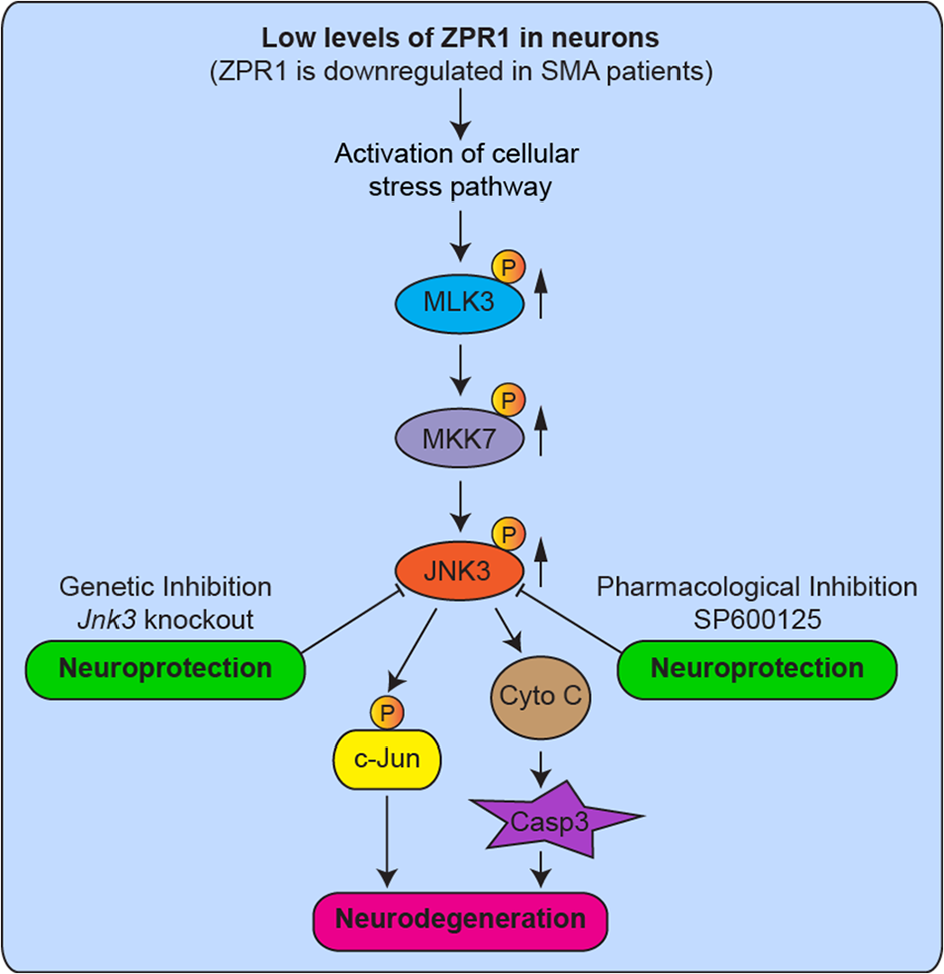
The molecular mechanism of ZPR1-dependent neurodegeneration. Graphical summary and the molecular mechanism of activation of the JNK signaling pathway in ZPR1-deficient neurons that mediates neurodegeneration. Patients with SMA express the low levels of ZPR1. Reduced levels of ZPR1 cause motor neuron degeneration and increase severity of disease in mice with SMA. SMA is characterized by degeneration of spinal motor neurons caused by chronic low levels of SMN. ZPR1 knockdown results in activation of the JNK signaling pathway MLK3 → MKK7 → JNK3 that mediates phosphorylation of c-Jun, release of Cytochrome C (Cyto C), and activation of caspase 3 (Casp3), which leads to axonal degeneration and caspase-mediated apoptotic death of neurons. Neuron-specific isoform JNK3 was identified to mediate degeneration of ZPR1-deficient neurons. Genetic inhibition of the JNK pathway by the Jnk3 gene knockout reduces/prevents degeneration of neurons. Pharmacologic inhibition of JNK using JNK inhibitor compound SP600125 reduces degeneration of ZPR1-deficient neurons and provide neuroprotection. Solid arrows (upward) show increase in phosphorylation and activation of MAP kinases. JNK indicates c-Jun NH2-terminal kinase; MAP, mitogen-activated protein; SMA, spinal muscular atrophy; SMN, survival motor neuron.
JNK pathway-mediated ZPR1-dependent neurodegeneration and its contribution to SMA pathogenesis
ZPR1 deficiency causes caspase-mediated cell death in cultured Zpr1 homozygous knockout mouse embryos (E3.5) and cultured motor neuron-like cells (NSC-34), which was prevented by the treatment with caspase inhibitor zVAD suggesting that ZPR1 deficiency may result in activation of caspase-mediated apoptosis/cell death. 3 Our current data demonstrate that ZPR1 deficiency indeed results in Cyto C-mediated cleavage and activation of Caspase 3 in primary cultured neurons and supports the preliminary published findings that ZPR1 deficiency may lead to caspase-mediated apoptotic cell death or degeneration of neurons.
The JNK signaling pathway plays a critical role in cell survival and death and implicated in the pathogenesis of human diseases, including cancer and neurodegenerative disorders. 32 The JNK signaling pathway is activated by extracellular stress signals, including growth factors, cytokines, and ultraviolet radiations. 24 JNK pathways have been shown to mediate caspase-dependent cell death and neurodegeneration.20,24,31,33,34 ZPR1 deficiency in neurons may cause intracellular stress that results in activation of intracellular MAPK cascade, which leads to JNK-mediated neurodegeneration. Activation of upstream kinases in the JNK cascade, MLK3 (MAP3K) and MKK7 (MAP2K) but not MKK4 (MAP2K) suggests that a specific signaling module MLK3 → MKK7 → JNK is activated in response to the loss of ZPR1 in CGN. It is possible that MLK3 activates MKK7 and MKK7 activates JNK and the specificity of activation is maintained by interaction of MLK3 and MKK7 with scaffold protein JNK-interacting protein 2 (JIP2) that forms a cascade module MLK3 → MKK7 → JNK. 25 Furthermore, our data demonstrate the activation (~25-fold) of neuron-specific isoform JNK3 in ZPR1-deficient neuron suggesting that a neuron-specific signaling cascade MLK3 → MKK7 → JNK3 mediates ZPR1-dependent neurodegeneration.
Patients with SMA express low levels of ZPR1.6,14,15 Activation of the JNK signaling pathway has been shown to mediate degeneration of the spinal cord motor neurons in SMA mice and the spinal cords of SMA patients. 20 Neurodegeneration in SMA is mediated by neuron-specific JNK3 isoform. 20 Genetic inactivation of Jnk3 gene in mice with SMA results in amelioration of SMA phenotype independent of alterations in SMN levels, including reduced loss of spinal motor neurons, increased overall growth and increased lifespan of mice with SMA. 20 Because ZPR1 is downregulated in SMA and our current data show that the low levels of ZPR1 result in the spinal cord motor neuron degeneration mediated by the JNK pathway, which suggest that the low levels of ZPR1 found in SMA patients may contribute to neurodegeneration associated with SMA pathogenesis. The downstream mechanism of ZPR1-dependent JNK activation is unclear. However, ZPR1 interacts with SMN and is required for accumulation of SMN in sub-nuclear bodies, including gems and CBs.3,6 ZPR1 colocalizes with SMN in gems and CBs. ZPR1 deficiency causes disruption of SMN containing nuclear bodies, which results in mislocalization of SMN and the loss of ZPR1-SMN complexes in the nucleus that may cause defects in the normal function of SMN leading to activation of the JNK-mediated cell death pathway and neurodegeneration in SMA.3,16,17
Activation of the JNK pathway in ZPR1-deficient neurons suggests that accumulation of intracellular stress may trigger cell death mechanisms that lead to neurodegeneration. It is possible that different types of intracellular stresses, including cellular toxicity, genomic instability, and reactive oxygen species (ROS) generated by ZPR1 and SMN deficiencies may contribute to activation of the JNK signaling pathway in SMA.20,29,31 Accumulation of DNA damage results in genomic instability (intra-cellular stress) and induces JNK-mediated apoptotic cell death.35-37 This is supported by recent findings that neurodegeneration in SMA is caused by accumulation of DNA damage leading to genomic instability. 22 Chronic low levels of SMN in SMA result in downregulation of Senataxin (SETX) and DNA-activated protein kinase-catalytic subunit (DNA-PKcs). SETX is an RNA-DNA helicase required for resolution of RNA-DNA hybrids (R-loops) formed during transcription. Senataxin deficiency results in accumulation of R-loops that cause DNA double-strand breaks (DSBs). Deficiency of DNA-PKcs impairs DSB repair leading to genomic instability.
In proliferating (dividing) cells, DNA damage is repaired by homologous recombination (HR) and non-homologous end joining (NHEJ). However, neurons (non-dividing cells) predominantly use NHEJ-mediated DNA repair, which relies on DNA-PKcs activity. Thus, DNA-PKcs deficiency results in inefficient NHEJ-mediated DNA repair resulting in accumulation of DNA damage that causes neurodegeneration in SMA. 22 Together, these findings that SMN deficiency causes DNA damage and JNK activation and the JNK pathway mediates apoptosis induced by DNA damage suggest that activation of JNK may be downstream of DNA damage associated with the pathogenesis of SMA.
Prevention of ZPR1-dependent neurodegeneration and therapeutic implications
The low levels of ZPR1 in patients may contribute to the severity and pathogenesis of SMA disease. This is supported by previous findings, which show that reduced Zpr1 gene dosage in mice with SMA increases the loss of spinal cord motor neurons, reduces growth, and decreases the lifespan of mice with SMA. 15 It is established that death in patients with SMA is caused by respiratory failure. 8 However, mechanisms that mediate respiratory distress in SMA are unclear. Recently, we have shown that ZPR1 is required for the survival of phrenic nerve neurons located in the cervical region C3-C5 of the spinal cord that regulates respiration. Spatiotemporal downregulation of ZPR1 in motor neurons causes degeneration of phrenic nerve motor neurons, which regulate respiration, and defects in innervation of the diaphragm leading to failure of synapse formation and respiration suggested that ZPR1 is critical for function of phrenic motor neurons. 18 Furthermore, analysis of ZPR1 levels in the spinal cord showed that ~50% downregulation of ZPR1 causes loss of phrenic motor neurons in the cervical region of mice with severe SMA. 18 These findings suggested that ZPR1 is critical for the normal function and survival of SMN-deficient neurons in mice with SMA. Therefore, prevention of ZPR1-dependent neurodegeneration may help reduce degeneration and ameliorate or rescue SMA phenotype.
In this study, we demonstrate that ZPR1-dependent neurodegeneration that is mediated by JNK3 is prevented by genetic and pharmacologic inhibitions of the JNK signaling pathway. Neurons derived from Jnk3–/– mice show reduced degeneration (~21%) compared with degeneration (~75%) of Jnk3+/+ neurons upon ZPR1 knockdown suggesting that JNK3 deficiency markedly increases survival (~54%) of neurons. These data are consistent with previous findings that the genetic deletion of Jnk3 reduced degeneration of SMN-deficient neurons in vitro and in vivo in mice with SMA. 20 Protection of ZPR1 or SMN-deficient neurons by knockout of Jnk3 (JNK3 deficiency) validates JNK3 as a potential therapeutic target to be exploited for developing a pharmacologic inhibition method using small drug compound. Treatment of ZPR1-deficient neurons with a broad-spectrum JNK inhibitor SP60012528,38 reduced degeneration (~31%) compared with degeneration (~75%) of DMSO-treated neurons suggesting that pharmacologic inhibition of JNK increases survival (~44%) of neurons. Comparison of genetic and pharmacologic methods shows higher neuroprotection (~10%) with genetic inhibition than the pharmacologic method. However, reduced neuroprotection (~10%) with pharmacologic inhibition might be because of the broad specificity and low efficacy of SP600125 toward JNK3. Nevertheless, these data suggest that pharmacologic JNK inhibition may be a viable therapeutic option to prevent or slow degeneration of ZPR1 and SMN-deficient neurons in SMA. A recent study showed pharmacologic inhibition of JNK using a specific JNK-inhibitor peptide reduced neurodegeneration and ameliorated SMA disease severity and increased the lifespan of SMAΔ7 mice similar to SMAΔ7 lacking Jnk3 (SMA-J3) mice.20,39 In conclusion, this study demonstrates that two independent methods, genetic and pharmacologic, for inhibition of JNK result in reduced degeneration of ZPR1-deficient neurons and suggest that JNK inhibition may be a viable strategy to prevent/slow neurodegeneration caused by the low levels of ZPR1 associated with the pathogenesis of SMA.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by research grants to L.G. from the US National Institutes of Health (R01 NS064224) and Muscular Dystrophy Association (MDA 480210). We thank Dr Zhanying Zhang for help with neuron culture, graduate student, Ms Deborah Aldrete for critically reading the manuscript, and Ms Marisol Ramirez for help with administrative assistance.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
LG conceived and received funding for this research project. XJ, AK and LG performed experiments. XJ, AK and LG organized and analyzed data, prepared figures and wrote/revised manuscript.