
Abstract
Most cases of spinal muscular atrophy are caused by functional loss of the survival of motor neuron 1 (SMN1) gene, while less than 5% of cases are attributed to genes other than SMN. Mutations in LMNA, the lamin A/C encoding gene, cause an adult form of spinal muscular atrophy (SMA), and in our recent work, we highlight a role for lamin A/C in SMN-related SMA pathways. Here, we discuss this apparent molecular crosstalk between different types of SMA in context with previous work, showing that dysregulation of proteins produced by other SMA-causing genes, including UBE1, GARS, and SETX, are also implicated in SMN-related SMA pathways. The perturbation of UBE1, GARS, and lamin A/C help explain mechanisms of tissue-specific pathology in SMA, and we propose Wnt/β-catenin signalling as a common molecular pathway on which they each converge. Therapeutic strategies directed at these proteins, or their convergent pathways, may therefore offer a new approach to targeting tissue-specific pathology in SMN-related SMA.
Spinal muscular atrophies (SMAs) are a heterogeneous group of neuromuscular disorders, clinically characterised by lower motor neuron loss, and muscle weakness and atrophy, but with varied age of symptom onset and severity of motor function impairments, and differential involvement of other organs. Spinal muscular atrophies are generally classified as either proximal SMA or distal SMA based on the limb region primarily affected by muscle weakness. Approximately 95% of SMA cases are caused by homozygous functional loss of the survival of motor neuron 1 (SMN1) gene, 1 resulting in insufficient levels of survival of motor neuron (SMN) protein. A minority (ie, <5%) of SMA cases involve genes other than SMN1, and to date, at least 30 different genes have been attributed to cases of non-SMN-related SMA (reviewed by Farrar and Kiernan 2 ). Intriguingly, experimental evidence has emerged from our recent work 3 and the work of others that proteins produced by several non-SMN-related SMA genes, including LMNA, 3 UBE1,4,5 GARS, 6 and SETX, 7 are implicated in SMN-related SMA pathways.
Mutations in LMNA, the lamin A/C encoding gene, cause a range of neuromuscular conditions with prominent cardiac involvement, including an adult form of SMA,8,9 and our recent study also proposes a role for lamin A/C in SMN-related SMA pathways. 3 Frequent reports of cardiac abnormalities in SMN-related SMA patients and mouse models of SMN-dependent SMA 10 prompted us to study the underlying molecular pathways using quantitative proteomics analysis. A key finding from this study was the identification of increased lamin A/C levels as a robust molecular phenotype in the heart of SMN-related SMA mice. 3 This increase would inevitably increase rigidity of nuclei leading to disrupted contractile activity in cardiomyocytes and thereby provide a mechanism to explain previous reports of morphological and functional cardiac defects in patients and SMA mice. 10 Lamin A/C dysregulation was also apparent in fibroblast cells from individuals with severe SMN-dependent SMA and in other tissues from SMN-dependent SMA mice, but with differing directions of expression change depending on the tissues examined. 3
A role for lamin A/C in SMN-dependent SMA is further strengthened by experiments in which we demonstrated a mechanistic link between lamin A/C and ubiquitin-like modifier activating enzyme 1 (UBA1) protein. 3 Mutations in the UBE1 gene, which encodes the UBA1 protein, cause a form of X-linked infantile SMA (SMAX2), 11 and involvement of UBA1 in SMN-dependent pathways has also been well characterised across several models of SMN-dependent SMA. UBA1 levels were reduced in mouse 4 and zebrafish models of SMN-dependent SMA, 5 and in induced pluripotent stem cell–derived motor neurons from individuals with severe SMN-related SMA.5,12 Pharmacologic or genetic suppression of UBA1/UBE1 phenocopied the SMA motor neuron phenotype in zebrafish, thus demonstrating that UBA1 contributes directly to SMN-related SMA disease pathways. 4 Systemic restoration of UBA1 levels increased motor performance in zebrafish and mouse models of SMA, as well as increased survival and improved systemic pathology in SMA mice. 5
The link between lamin A/C and UBA1 is likely to involve β-catenin, as both lamin A/C 13 and UBA1 are implicated in regulating β-catenin signalling, 4 and β-catenin itself contributes to SMN-dependent SMA pathways. 4 Defective Wnt/β-catenin signalling, for example, was shown to contribute to the pathology of dilated cardiomyopathy caused by mutations in the LMNA gene. 13 Decreased expression of several components of Wnt/β-catenin pathway, including β-catenin, was identified in the heart from a mouse model of LMNA cardiomyopathy, and pharmacologic activation of Wnt/β-catenin signalling improved cardiac pathology in these mice. 13 In a separate study, lamin A/C overexpression increased nuclear levels of β-catenin and activated the Wnt signalling pathway to promote osteoblast differentiation. 14 Our recent study 3 further expanded on the link between lamin A/C and β-catenin by demonstrating that they interact in mouse heart extracts under normal physiological conditions. UBA1, on the other hand, controls the stability of β-catenin through the canonical ubiquitin-proteasome pathway, and deficiency in UBA1 protein levels leads to β-catenin accumulation and neuromuscular pathology in SMN-dependent SMA. 4 Pharmacological inhibition of β-catenin signalling, using quercetin, ameliorated neuromuscular pathology in Drosophila, zebrafish, and mouse models of SMN-dependent SMA. 4
Mutations in GARS, the gene encoding glycine-tRNA synthetase (GARS), are responsible for Charcot-Marie-Tooth disease Type 2D (CMT2D), typically characterised by sensory impairment, but also cases of both childhood and adult forms of distal SMA, termed distal SMA type V. 2 Studies of GARS, which was found to be another downstream target of UBA1 pathways, provided further insights into the fundamental molecular mechanisms driving pathology in SMN-dependent SMA tissues. 7 Increased expression of GARS in spinal cords from a mouse model of severe SMN-dependent SMA was restricted to sensory neurons and increasing the expression of UBA1 was sufficient to restore GARS levels and correct sensory neuron defects in severe SMN-dependent SMA mice. 7 UBA1/GARS pathway dysregulation is therefore likely to be responsible, at least in part, for disrupted sensory neuron fate and altered sensory-motor connectivity in these mice. 7 The amino acid-tRNA synthetases (ARS), of which GARS is one, form a multi-synthetase complex with 3 scaffold proteins called aminoacyl-tRNA synthetase-interacting multifunctional proteins (AIMP1, AIMP2, and AIMP3). A study of the intestinal epithelium and tumorigenesis found evidence of enhanced Wnt/β-catenin signalling in mice harbouring a hemizygous deletion of AIMP2, 15 which therefore indirectly implicates ARS proteins in β-catenin signalling pathways too. Further work would be required to confirm and verify this across other cells and tissues, but it does, nonetheless, raise the possibility that convergence on Wnt/β-catenin signalling pathways could well be a common mechanistic link between UBA1, lamin A/C, and GARS in SMN-dependent SMA (Figure 1).
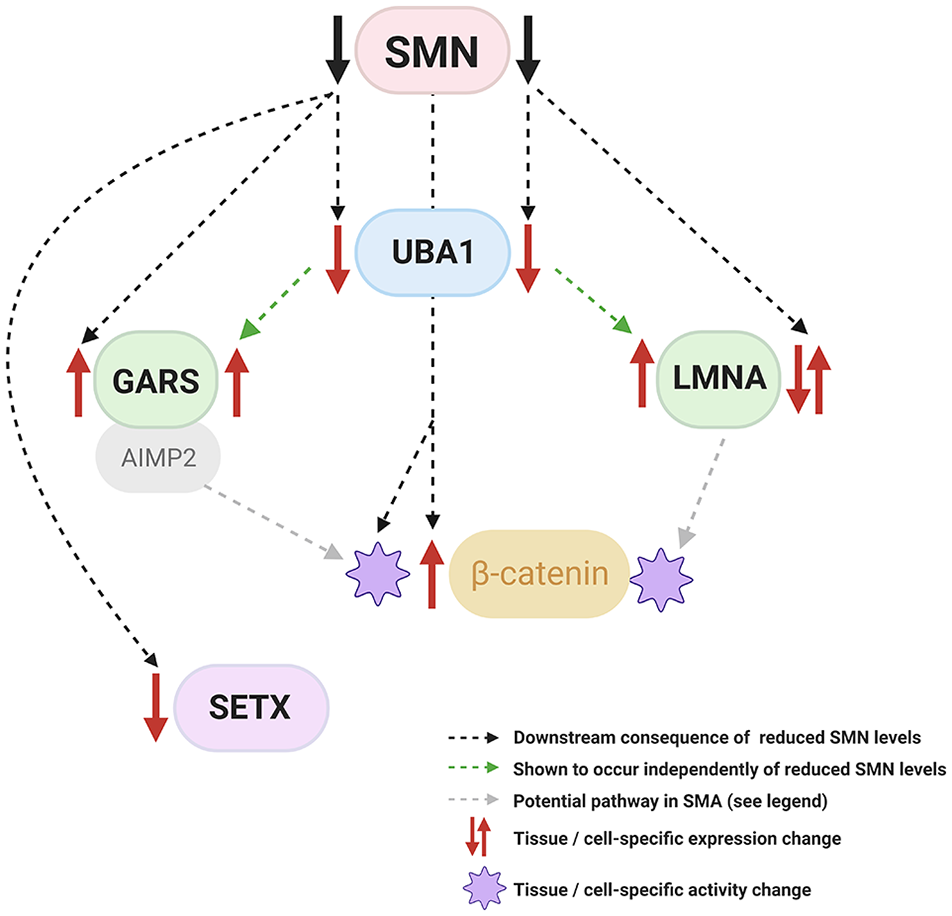
Proteins produced by several non-SMN-related SMA genes are implicated in SMN-related SMA pathways. Alterations to lamin A/C, UBA1, GARS, and SETX have been reported as downstream consequences of reduced SMN. Further work has shown that UBA1 is able to modulate lamin A/C and GARS expression independently of SMN. UBA1 converges on β-catenin signalling pathways in SMN-related SMA, and evidence from studies unrelated to SMA also implicates GARS and lamin A/C in the modulation of Wnt/ β-catenin signalling. Image created with BioRender.com.
Mutations in the SETX gene, which encodes the (probable helicase) senataxin protein, can cause several conditions including ataxia with oculomotor apraxia type 2, a type of amyotrophic lateral sclerosis, and an autosomal dominant form of proximal spinal muscular atrophy (ADSMA). 16 Like SMN, senataxin is involved with maintaining RNA transcriptome homeostasis 16 and is another non-SMN-related SMA candidate implicated in SMN-related SMA pathways. Decreased senataxin protein levels were identified in fibroblasts and spinal cords from individuals with SMN-dependent SMA, cultured spinal cord neurons from a mouse model of SMN-dependent SMA, and in SMN-deficient HeLa cells. 7 Consistent with one of its known functions, the decreased expression of senataxin in vitro coincided with increased accumulation of R-loops and double-strand breaks (DSBs). 7 Restoration of SMN in vitro restored senataxin levels and decreased R-loop accumulation – and overexpression of senataxin alone was sufficient to decrease R-loop accumulation – in SMN-related SMA patient fibroblasts and cultured mouse spinal cord neurons. 7 This evidence, therefore, suggests that senataxin is, at least partially, responsible for genomic instability in SMN-related SMA in vitro, most likely via its helicase activity. Mutations in the gene encoding immunoglobulin mu DNA binding protein 2 (IGHMBP2) – a protein with high sequence homology to the helicase domain of senataxin – cause a type of severe SMA with respiratory distress, 16 which further highlights the importance of helicase activity in SMA disease pathways.
There is no cure for any type of SMA but significant progress has been made recently in the development of therapies aimed at raising full-length SMN protein levels for SMN-related SMA. 17 Nusinersen (Spinraza™), an antisense oligonucleotide drug, is now widely available for children and young adults with SMA, and most recently, Zolgensma™, an adeno-associated virus-based gene replacement therapy, was given approval by the Food and Drug Administration (FDA) for the treatment of SMA children in the United States under 2 years of age. Despite these incredible advances, both strategies are extremely expensive, neither show complete efficiency, 17 and the long-term outcomes remain unknown. In addition, where neuronal tissue is the only target of the treatment (ie in the case of Spinraza™), peripheral pathologies may still be a concern. Consequently, there is a need for a new generation of SMA therapies that could, in combination with SMN-targeted therapy, offer maximal benefit to SMN-related SMA patients. Recent preclinical work has positioned four non-SMN-related SMA proteins as key players in SMN-related SMA disease pathways, some of which help explain the mechanisms of tissue-specific pathology in SMN-related SMA. Therapeutic strategies directed at these proteins, or pathways on which they converge, may therefore offer a new approach to targeting tissue-specific pathology in SMN-related SMA.
Footnotes
Acknowledgements
We are grateful to H.K. Shorrock, H. Allardyce, E.L. Wilson, I. Holt, S.A. Synowsky, S.L. Shirran, S.H. Parson, and T.H. Gillingwater for their collaboration in the original study. In instances where review articles are cited above, we would also wish to acknowledge authors of original work that we were unable to cite directly due to space constraints.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Spinal muscular atrophy research in H.R.F.’s laboratory is currently supported by Sparks and Great Ormond Street Hospital Children’s Charity (grant number V5018).
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
DS and HRF wrote the manuscript.