
Abstract
Indoleamine-2,3-dioxygenase (IDO) degrades the essential amino acid tryptophan resulting in tryptophan depletion and the accumulation of catabolites such as kynurenine. The expression/activity of IDO in various cells, including macrophages and dendritic cells, results in an inhibition of T-cell responses in a number of situations, such as toward allogeneic fetuses and tissue grafts. Psoriasis is an immune-mediated skin disease involving T cells; kynureninase and its generation of catabolites downstream of IDO are reported to play an important role in this disease. We hypothesized that mice lacking the IDO1 gene would exhibit a hyperactive immune response and an exacerbation of skin lesions in the imiquimod-induced mouse model of psoriasis. Littermate wild-type and IDO1-knockout mice were treated with imiquimod for 5 days, and the severity of psoriasiform skin lesions assessed using the psoriasis area and severity index (PASI), ear edema measured using a digital caliper, and thickness of the epidermis determined by histology. Expression of pro-inflammatory mediators and tryptophan-metabolizing enzymes was monitored using quantitative RT-PCR. Imiquimod increased ear edema, PASI scores, and epidermal thickness in both WT and IDO1 knockout mice; however, there were no differences observed between the 2 genotypes. There were also no differences in imiquimod’s induction of skin inflammatory mediators, indicating no effect of IDO1 gene loss in this psoriasis model. Although these data suggest a lack of involvement of IDO1 in psoriatic skin inflammation, other possible mechanisms, such as compensatory changes in other pathways and the involvement of the IDO2 isoform, must also be considered.
Keywords
Introduction
Indoleamine 2,3-dioxygenase (IDO) is an enzyme that catalyzes the first and rate-limiting step in the degradation of the essential amino acid tryptophan to kynurenine. Although this reaction can also be performed constitutively by liver tryptophan 2,3-dioxygenase (TDO), IDO can be induced by various inflammatory mediators and has been found to play a key role in regulating the responsiveness of the immune system. In pioneering studies Munn and Mellor showed that IDO activity in antigen-presenting cells could activate regulatory T cells (Tregs) and inhibit the proliferation of effector T cells thereby suppressing immune responses.1,2 This mechanism appears to be involved in a number of processes, including immune tolerance in pregnancy, cancer, and transplantation of tissue grafts (reviewed in Mbongue et al 1 and Munn and Mellor 3 ). Indeed, Munn et al 4 demonstrated that inhibition of IDO with 1-methyltryptophan induces T cell-mediated rejection of allogeneic but not syngeneic offspring in pregnant mice. Immune suppression by IDO is known to occur through at least 2 mechanisms: (1) kynurenine produced by IDO can suppress T cell responses and stimulate Treg differentiation through the aryl hydrocarbon receptor (reviewed in Mbongue et al 1 and Munn and Mellor 3 ). And (2) depletion of tryptophan results in activation of the kinase general control nonderepressible-2 (Gcn2), which can promote the differentiation of naïve T cells into Tregs and stimulate their activity (reviewed in Mellor et al 5 ); Gcn2 can also inhibit proliferation of T cells and their differentiation to Th17 cells. Therefore, IDO is thought to be a key immunomodulatory enzyme and the tryptophan-kynurenine degradation pathway an important contributor to disorders of immune tolerance including autoimmune disease and tissue graft rejection.
Accumulating evidence indicates the importance of the Th17 immune response in the skin disease psoriasis. 6 Psoriasis is a common, immune-mediated skin condition characterized by epidermal keratinocyte hyperproliferation and abnormal differentiation, inflammation, immune system activation, and skin infiltration of immune cells. 7 The resulting skin lesions may be pruritic and are esthetically displeasing, such that patients with psoriasis often report a poor quality of life.8,9 In addition, the chronic inflammation associated with the disease can predispose people with psoriasis to other co-morbidities,10 -12 such as cardiovascular issues. Analyses of the gene expression patterns in lesional and non-lesional psoriatic skin and in normal skin have identified a number of potential players, including the Th17 pathway and its associated cytokines, in psoriasis. In addition, IDO1 mRNA and protein levels have been shown to be increased in lesional psoriatic skin,13,14 suggesting the likely importance of this pathway in the disease. Of note, Harden et al compared 3 psoriasis datasets: (1) “psoriasis classifier” genes, which can correctly distinguish between lesional and non-lesional skin, (2) psoriasis methylation genes that show altered methylation patterns in psoriasis, and (3) “molecular scar” genes, which are slightly upregulated in non-lesional compared to normal skin after treatment of psoriasis with anti-tumor necrosis factor-α (TNFα) therapy. Their analysis identified a single gene found in all 3 sets, kynureninase, a kynurenine-degrading enzyme downstream of IDO. 13 These authors also demonstrated an ability of downstream kynurenine catabolites, such as 3-hydroxyathranilic acid and quinolinic acid (Figure 1), to stimulate keratinocyte inflammatory gene expression. 13

The tryptophan-kynurenine catabolic pathway. Shown is the catabolism of tryptophan by indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO) to kynurenine through the N-formylkynurenine intermediate. Kynurenine can then be catabolized to anthranilic acid by kynureninase (KYNU), to 3-hydroxykynurenine (3-OH-Kyn) by kynurenine 3-hydroxylase (K3H) or to kynurenic acid by kynurenine aminotransferase (KAT). Further catabolism to xanthurenic acid, 3-hydroxyanthranilic acid (3-OH-AA), and quinolinic acid (through 3-hydroxyanthranilate 3,4-dioxygenase, 3HAO) is also illustrated.
Based on the data suggesting the importance of IDO in regulating the immune system (reviewed in Munn and Mellor 2 ), we hypothesized that an immune-mediated, inflammatory skin condition like psoriasis would be exacerbated in a mouse genetically lacking the IDO1 gene. However, it should be noted that there are 2 isoforms of IDO, IDO1 and IDO2, both of which may play a role in regulating the immune system. Indeed, Fujii et al 15 demonstrated an exacerbation of the psoriasis-like dermatitis phenotype induced by imiquimod (IMQ) in IDO2 knockout mice, suggesting a potentially important role for this IDO isoform in skin. We used IDO1 knockout (KO) mice and their wild-type (WT) littermates to determine the effect of IDO1 gene ablation on IMQ-induced psoriasis-like dermatitis in a mouse model of psoriasis. 16 We selected this particular model because the skin lesions developed in response to IMQ exposure: (a) exhibit similar characteristics to those observed in psoriatic lesions in humans, 16 with parakeratosis, agranulosis, and microabscess formation, (b) involve key cytokines, such as the IL17 pathway, as in psoriasis, 16 and (c) exhibit corresponding gene expression patterns to those found in psoriatic lesions. 17 In addition, (d) this model is rapidly generated in genetically engineered mice, such as the IDO1 knockout mice, without the need for additional breeding. Furthermore, and perhaps more importantly, in some patients who receive IMQ to treat genital warts, actinic keratosis or inoperable superficial basal cell carcinoma, IMQ can initiate or exacerbate psoriasis. 16 Therefore, IMQ-induced dermatitis in mice seems to be a good model for psoriasis and should provide insight into the possible role of IDO1 in this skin disease.
Methods
Animal experiments
IDO1 KO and WT littermates were bred by crossing IDO1 KO mice on the C57BL/6J strain background, graciously provided by Dr. David Munn (Augusta University, Augusta, GA, USA), with C57BL/6J wild-type (WT) mice obtained from Jackson Laboratory (Bar Harbor, ME, USA) to generate heterozygotes. The heterozygotes were then crossed to produce IDO1 KO and WT littermates for analysis to ensure that the IDO1 KO was compared to the appropriate WT control. The IMQ-induced mouse model of psoriasis was then generated as described in van der Fits et al 16 and Choudhary et al. 18 Briefly, the backs of male IDO1 KO and WT mice, between the ages of 8 and 10 weeks, were shaved and depilated using Veet® (obtained from a local pharmacy) under isoflurane anesthesia. Two days later the development of psoriasiform lesions was initiated by topically applying 62.5 mg of Aldara™ cream (Meda Pharmaceuticals, Somerset, NJ, USA) or a vehicle (Vaseline, obtained from a local pharmacy) to the back and right ear of the mice. Vehicle or Aldara® were applied daily for an additional 4 days, with skin lesions monitored daily. On the sixth day mice were sacrificed and the ear inflammation monitored using a digital caliper to measure thickness and an AX26 DeltaRange microbalance (Mettler Toledo, Columbus, OH, USA) to determine the weight of a 4 mm punch biopsy of the ears. A portion of back skin was fixed in formalin for paraffin embedding, sectioning, and staining with H&E to monitor epidermal thickness (see below). From a subset of animals (3 mice per each control group and 5 mice per each imiquimod-treated group), back skin was also harvested and flash-frozen for subsequent homogenization, RNA isolation, and quantitative RT-PCR analysis of gene expression (see below). All animal procedures were approved by the Augusta University Institutional Animal Care and Use Committee (Protocol #2015-0725).
Histology and measurement of epidermal thickness
Sections (5 µm) were cut from formalin-fixed, paraffin-embedded skin blocks. Slides were processed and stained with hematoxylin and eosin (H&E) via standard histological procedures. Photomicrographs of the epidermis of each section were taken randomly and analyzed to determine epidermal thickness using ImageJ (National Institutes of Health, Bethesda, MD, USA) by 3 observers who were blinded to the sample identity, as described previously. 18
RNA isolation
Frozen skin tissue was homogenized using a mortar and pestle under liquid nitrogen. The powder was solubilized and RNA isolated using TRIzol™ (ThermoFisher Scientific, Waltham, MA, USA) as per the manufacturer’s protocol and as described previously. 18 The total RNA was checked for quality and quantified using a Nanodrop instrument (ThermoFisher Scientific). One microgram of total RNA was used to generate complementary DNA using ABI High-Capacity cDNA Reverse Transcription kits (ThermoFisher Scientific). Quantitative PCR was then performed in a StepOnePlus (ThermoFisher Scientific) instrument using Taqman primer-probe sets and Taqman reagents (ThermoFisher Scientific) as per the supplier’s instructions. The primer-probe sets used were: mouse Ido1 (Mm00492590_m1), Il1b (Mm00434228_m1), Il6 (Mm00446190_m1), Tnfα (Mm00443258_m1), kynureninase (Mm00551012_m1), and Ido2 (Mm00524210_m1). Mouse Gapdh gene (Mm99999915_g1) was used as an endogenous housekeeping gene for delta-delta Ct analysis.
Mass spectrometric analysis of serum tryptophan and kynurenine levels
Serum samples, obtained by submandibular blood draw from 4-month-old IDO1 KO and WT mice, were processed by adding 120 µL of methanol to 20 µL of mouse serum and vortexing for 1 minute at room temperature, followed by centrifugation at 16 000×g at 4°C for 15 minutes. Supernatants (80 µL) were transferred to sample vials and mixed with 40 µL of water containing 0.1% formic acid. Tryptophan and kynurenine were then separated and quantified by LC-MS analysis using a Phenomenex Kinetex C18 column (100 × 2.1 mm, 1.7 µm) with a Shimadzu Nexera UHPLC system. A gradient of 5% to 40% acetonitrile (containing 0.1% formic acid) was performed over 6 minutes at a flow rate of 0.2 mL/minute and a column temperature of 40°C. The effluent was ionized using positive ion electrospray on a ThermoScientific TSQ Quantiva triple quadrupole mass spectrometer with the following instrument settings: ion spray voltage 3500V, sheath gas 35, aux gas 10, ion transfer tube temperature 325, vaporizer temperature 250, and Q1 and Q3 resolution 0.7 FWHM. The optimal fragments (Q3), collision energy (V), and RF lens (V) were determined using purchased standards and the parameters listed below:

After LC-MS analysis, the raw data were imported into Skyline software (V20.0) to calculate the integrated peak areas of the transitions for the standards and samples.
Statistical analysis
Differences were determined using one-way analysis of variance (ANOVA) followed by Newman-Keuls post-hoc tests on the number of mice (ie, the n) indicated in each figure legend; statistical significance was assigned at P < .05. Statistical analyses were performed using GraphPad Prism (San Diego, CA, USA). The results of the PASI scores over time of treatment were analyzed using a mixed model repeated measures ANOVA with a Bonferroni correction for comparisons of IMQ and vehicle for each day of treatment using SAS© V9.4 (SAS Institute, Inc., Cary, NC).
Results
Imiquimod induces the development of psoriasiform lesions to a similar extent in WT and IDO1 KO mice
IMQ was applied topically to the skin of WT and IDO1 KO daily for 5 days and the skin lesions were photographed. These skin lesions were evaluated by a practicing dermatologist blinded to the treatment in terms of their area, erythema, scaling, and thickening to give psoriasis area and severity index (PASI) scores. Figure 2A shows representative mice of each genotype treated with vehicle or IMQ on day 6, with mean PASI scores for days 3 through 6 shown in Figure 2B. PASI scores on day 6 are also shown (Figure 2C). As illustrated, IMQ induces psoriasiform lesion development over time and to a similar degree in WT and IDO1 KO mouse littermates.

Loss of IDO1 did not affect the imiquimod-induced development of psoriasiform skin lesions in the imiquimod-induced mouse model of psoriasis. (A) Wild-type (WT) and IDO1 knockout (KO) mice were treated daily for 5 days with vehicle (control, Ct) or imiquimod (IMQ) on their shaved back and right ear as described in Methods. Psoriasis area and severity index (PASI) scores were estimated from photographs of the back skin by a practicing dermatologist in a blinded fashion for (B) days 3 through 6 (the day of sacrifice) or (C) for day 6 only. Data are presented as the means ± SEM of 8 mice per each control group and 10 mice per each IMQ group; ***P < .001 versus vehicle-treated mice. In panel B there was a significant interaction of treatment with time as indicated; ***P < .001 versus vehicle treatment.
Imiquimod induces a similar degree of ear edema in WT and IDO1 KO mice
IMQ-induced ear inflammation was also monitored on day 6 by measuring the differences in ear thickness between the treated (right) and untreated (left) ear using a digital caliper. Figure 3A demonstrates that IMQ application resulted in an increase in ear thickness in both WT and IDO1 KO mice, but this increase was not different between the 2 genotypes. Ear swelling measured using the weight of a 4 mm punch biopsy of the treated ear (in comparison to the untreated ear) showed a similar effect: IMQ application increased ear weights in both genotypes to a degree that was not statistically different (Figure 3B).

Loss of IDO1 did not affect ear swelling in response to imiquimod, as measured by ear thickness or weight. Mice were treated daily for 5 days with vehicle or IMQ on their shaved back and right ear as described in Methods. Immediately after sacrifice on the sixth day (A) ear thickness was measured using a digital caliper and (B) ear weight (of a 4 mm punch biopsy) was determined. Data are presented as the means ± SEM of 8 mice per each control group and 10 mice per each IMQ group; *P < .05, ***P < .001 versus the vehicle-treated control.
Imiquimod-increased epidermal thickness was not statistically different in the WT and IDO1 KO mice
Sections of fixed back skin were stained with H&E and epidermal thickness determined from random photomicrographs by observers that were blinded to the treatment/genotype. Representative sections for a mouse from each group are shown in Figure 4A. Cumulative results from all mice in each group are shown in Figure 4B, from which it is clear that IMQ induced an increase in epidermal thickness as expected and to a similar degree in WT and IDO1 KO mice.

Loss of IDO1 did not affect the imiquimod-induced increase in epidermal thickness. Mice were treated daily for 5 days with vehicle or IMQ on their shaved back and right ear as described in Methods. (A) After sacrifice, back skin was harvested and fixed in formalin for H&E staining as described in Methods. (B) Data are presented as the means ± SEM of 8 mice per each control group and 10 mice per each IMQ group; ***P < .001 versus the respective WT and IDO1 KO control values; ns, not significant.
IDO1 expression was undetectable in IDO KO mice, and imiquimod increased inflammatory mediator expression to a similar extent in WT and IDO KO mouse skin
Using quantitative RT-PCR it was first verified that the IDO1 KO mice expressed no IDO1; indeed, IDO1 mRNA levels were undetectable in the IDO1 KO mouse skin (Figure 5A). IMQ induced the expression of the inflammatory mediators examined: interleukin (IL)-1β, IL-6 and TNFα. However, there was no difference in the degree of stimulation of the expression of these mediators in the WT versus the IDO1 KO mouse skin (Figure 5B-D). Based on the report from Harden et al 13 indicating the importance of the downstream enzyme kynureninase in psoriasis and the study of Fujii et al 15 demonstrating the key role of IDO2 in the IMQ-induced mouse model of psoriasis, the expression of kynureninase and IDO2 in the skin from each mouse was also determined. Kynureninase expression was elevated in the IDO1 KO control (vehicle-treated) mice but was suppressed to WT levels upon treatment with IMQ. On the other hand, in WT mouse skin IMQ had no effect on kynureninase expression (Figure 5E). In terms of IDO2, there were no differences in mRNA levels between the WT and IDO KO mice but IMQ increased IDO2 expression (Figure 5F).
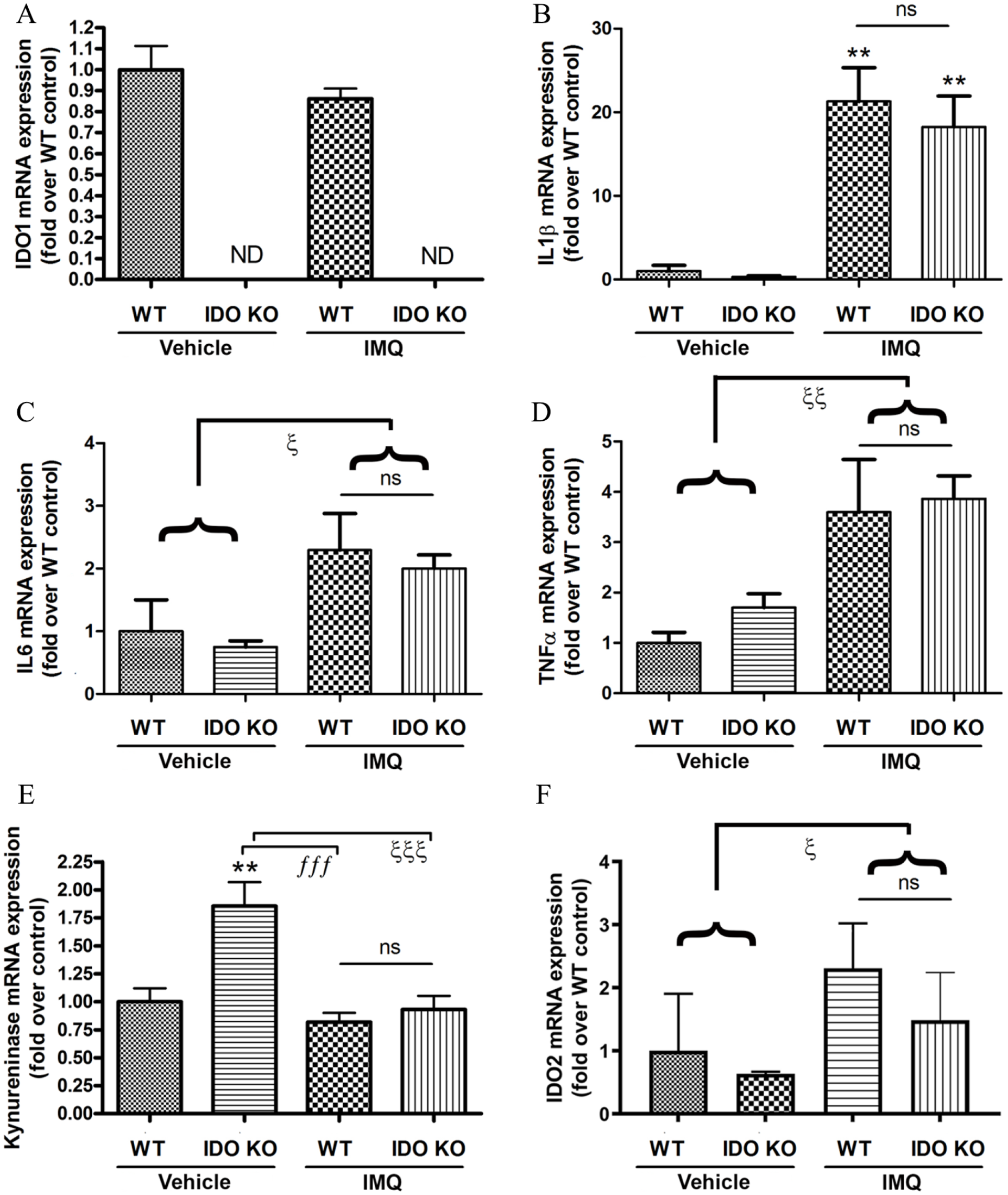
Loss of IDO1 did not affect the imiquimod-induced increase in inflammatory mediator expression in the skin. RNA isolated from the back skin of a subset of mice subjected to the indicated treatments as described in Methods was analyzed by quantitative RT-PCR for the expression of (A) IDO1, (B) IL1β, (C) IL6, (D) TNFα, (E) kynureninase, and (F) IDO2. Data represent the means ± SEM of 3 mice per each control group and 5 mice per each IMQ group. In the case of IL6, TNFα, and IDO2, there were no statistically significant differences detected by ANOVA; therefore, the control values and the IMQ-treated values for both genotypes were combined and analyzed by Student’s t-tests; **P < .01 versus the WT control; fffP < .001, P < .05, P < .01, P < .001 as indicated; ND, not detected; ns, not significant.
To determine whether similar changes in IDO2 were observed in a tissue other than the skin, kidneys were collected from some of the mice and RNA isolated. As in skin, IDO1 mRNA levels were undetectable in the IDO1 knockout mice (Figure 6A), confirming the global gene deletion in the IDO1 knockout mice. However, in contrast to skin, basal IDO2 mRNA levels were significantly and markedly decreased in the IDO1 knockout mice (Figure 6B). (Please note that IDO2 expression in WT mice seemed to be higher in kidney than in skin, with a cycle threshold of 31-32, compared to ~33-35.) Also, unlike in skin, kynureninase expression was not altered in the kidneys of the knockout mice compared to the wild-type animals (Figure 6C). This result suggests that kynurenine metabolites might have different roles in the skin versus the kidney. To verify that IDO1 gene ablation resulted in reduced IDO activity, we measured the basal serum levels of tryptophan and kynurenine in 4-month-old wild-type and IDO1 knockout mice by mass spectrometric analysis. Serum kynurenine levels were significantly reduced by about half in the knockout mice in comparison to wild-type controls (Figure 6E), while tryptophan levels were essentially unchanged (Figure 6D). These changes resulted in an approximately 2-fold greater tryptophan to kynurenine ratio in the serum of the IDO1 KO mice compared to the WT controls (Figure 6F). Therefore, the data support the idea that IDO1 activity contributes significantly to basal serum kynurenine levels and further that IDO1 global gene deletion leads to decreased indoleamine 2,3-dioxygenase activity.

Loss of IDO1 resulted in down-regulation of IDO2 and no change in kynureninase expression in the kidney, as well as decreased serum kynurenine levels and an increased tryptophan/kynurenine ratio. RNA isolated from the kidneys of a subset of mice treated with vehicle was analyzed by quantitative RT-PCR for the expression of (A) IDO1, (B) kynureninase and (C) IDO2. Data represent the means ± SEM of 3 to 5 mice per group. Blood was collected from the cheek pouch of a separate group of 4-month-old mice and serum (D) tryptophan and (E) kynurenine levels determined by mass spectrometry as described in Materials and Methods. Panel (F) illustrates the data for the tryptophan/kynurenine ratio for each mouse. Data represent the means ± SEM of 5 mice per group; **P < .01, ***P < .001.
Discussion
We wished to examine the role of IDO1 in an experimental mouse model of psoriasis based on the recognized involvement of IDO in regulating the immune system, and the known role of the immune system in psoriasis (reviewed in Mellor et al 5 and Helwa et al 7 ), as well as the data suggesting the importance of the tryptophan-kynurenine pathway in psoriasis. 13 We selected the IMQ-induced mouse model of psoriasis for several reasons as discussed in the Introduction. These include the fact that the use of IMQ in humans to treat certain skin diseases induces psoriasis in some individuals (reviewed in Flutter et al 19 ), suggesting a common mechanism of lesion development in mouse and human.
We hypothesized that IDO1 knockout mice would show exacerbation of psoriasiform lesion development, based on evidence in the literature. Thus, IDO1-overexpressing fibroblasts have been demonstrated to suppress the development of skin lesions in this same IMQ-induced mouse model of psoriasis. 20 In addition, Kim et al 21 determined that manipulations that increase reactive oxygen species result in the amelioration of psoriasiform lesion development in response to IMQ by upregulating IDO expression and enhancing Treg function. Furthermore, IDO, possibly IDO1, is upregulated in psoriasis lesions.13,14 Nevertheless, genes encoding other enzymes in the IDO pathway (eg, kynureninase) are also induced in psoriatic lesions, and Harden et al 13 actually proposed that kynurenine metabolites, such as 3-hydroxyanthranilic acid and quinolinic acid generated by kynureninase-mediated metabolism of IDO-generated kynurenine, may be responsible for the abnormalities observed in psoriasis. Therefore, it was conceivable that loss of the IDO1 gene would result in reduced levels of these metabolites and amelioration of the skin lesions. In control IDO1 KO mouse skin upregulation of kynureninase expression was observed, implying a possible attempt to restore levels of these metabolites by enhancing kynureninase activity in order to compensate for reduced IDO1 kynurenine metabolism. (Since the liver enzyme TDO can also produce kynurenine, kynurenine is still present, although at lower levels, as shown in Figure 6, in the sera of IDO1 knockout mice.)
Interestingly, IDO1 gene ablation neither exacerbated nor ameliorated IMQ-induced skin lesions. Although this result might be interpreted as indicating a lack of involvement of IDO1 in psoriasis, it is possible that these competing effects, reduced suppression of inflammation because of impaired Treg generation/function versus decreased generation of pro-inflammatory kynurenine metabolites, essentially counterbalanced each other to result in no difference in the psoriasiform phenotype. Alternatively, it seems likely that IDO2 rather than IDO1 is the key kynurenine-generating enzyme in skin. Indeed, Fujii et al 15 found that IDO2 was expressed in human epidermis and mouse ear skin and was upregulated in psoriasis and IMQ-induced psoriasisform skin lesions, respectively. IDO1 was reportedly not expressed in these tissues, in contrast to our finding of IDO1 expression in mouse back skin, albeit at low levels (a cycle threshold of ~33-35). The reason for this discrepancy is unclear but may be related to sex (male in ours vs female in the other study) or skin location (dorsum in ours vs ear). Consistent with the results of Fujii et al, 15 our results showed an increase in IDO2 mRNA levels in IMQ-treated mice, although as with IDO1, expression was low (a cycle threshold of ~33-36). Also in line with our results, Fujii et al 15 showed that a lack of IDO1 in knockout mice had little effect on IMQ-stimulated increases in ear erythema and scaling and epidermal thickness at 7 days, although IDO1 knockout mice were observed to exhibit enhanced changes in ear thickness initially upon IMQ treatment (days 2-4). In IDO2 knockout mice, on the other hand, IMQ-induced psoriasis-like dermatitis was worse than in the wild-type mice; thus, the IDO2 knockout mice exhibited greater increases in ear erythema, scaling and thickness as well as increased epidermal thickness and TNFα, IL-23p19 and IL-17a expression at 7 days in comparison with the wild-type animals. 15 Thus, IDO2, rather than IDO1, seems to be involved in psoriasis. However, the fact that IDO2 knockout mice show worse psoriasis-like skin lesions 15 suggests that IDO2 normally suppresses the disease phenotype. Therefore, it is perhaps not unexpected that IDO2 expression might be upregulated upon exposure of the skin to IMQ in an attempt to inhibit lesion development. However, this upregulation should result in increased kynurenine and its metabolites, which, based on the data of Harden et al, 13 should enhance keratinocyte proliferation and other psoriasis-like skin changes, although it is possible that there are species differences in the involvement of the kynurenine pathway in skin biology. Therefore, further investigation is required to determine the exact role of this pathway in the skin under normal and disease conditions.
In conclusion, our results show that IDO1 KO mice developed psoriasiform lesions in the IMQ-induced mouse model similarly to their WT littermates. Although this result could be interpreted to mean that there is no role for IDO1 in psoriasis, there are limitations to the study. Thus, it is possible that pro- and anti-psoriatic effects of IDO1 counterbalance each other. In addition, it should be noted that the IMQ model represents an acute model of psoriasis, and it is possible that a more chronic stimulation of the immune system would be required to unmask the importance of IDO1. Alternatively, compensatory mechanisms may be initiated to minimize the effect of loss of the IDO1 gene. Indeed, we detected upregulation of kynureninase gene expression in the control IDO1 KO mice, and the family member IDO2 was up-regulated with IMQ application; IDO2 is not redundant with IDO1, although it is much less well-studied. 2 Finally, the KO mice may have other, as yet unknown, compensatory effects that mitigate the lack of IDO1. In addition, since psoriasis is an inflammatory skin disease involving the innate and adaptive immune system as well as keratinocytes 22 (and possibly the vasculature and nervous system, eg,23,24), it is possible that IDO1 loss could induce contrasting (and counterbalancing) effects in different tissues. This idea is supported by the differences observed in skin versus kidney expression of the tryptophan-metabolizing enzymes IDO2 and kynureninase. Thus, additional studies using cell-specific IDO1 and IDO2 KO mice, as well as in humans, seem warranted to determine whether this enzyme and the kynurenine pathway contribute in any way to psoriasis manifestations.
Footnotes
Acknowledgements
Author’s Note
The contents of this article do not represent the views of the Department of Veterans Affairs or the United States Government.
Author Contributions
VC, CMI and WBB conceived of and designed the study; VC, EA, RU, SCS, SH, XC, JX, MC and DLI acquired the data; VC, RU, SCS, MC and WBB analyzed and interpreted the data; VC, EA and WBB drafted the manuscript and all authors revised and approved its submission.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the National Institutes of Health/National Institute on Aging (#AG036675 to CMI) and the Veterans Administration (VA Merit #CX001357 to WBB).