
Abstract
Nicotinamide, the amide form of vitamin B3 (niacin), has long been associated with neuronal development, survival, and function in the central nervous system (CNS), being implicated in both neuronal death and neuroprotection. Here, we summarise a body of research investigating the role of nicotinamide in neuronal health within the CNS, with a focus on studies that have shown a neuroprotective effect. Nicotinamide appears to play a role in protecting neurons from traumatic injury, ischaemia, and stroke, as well as being implicated in 3 key neurodegenerative conditions: Alzheimer’s, Parkinson’s, and Huntington’s diseases. A key factor is the bioavailability of nicotinamide, with low concentrations leading to neurological deficits and dementia and high levels potentially causing neurotoxicity. Finally, nicotinamide’s potential mechanisms of action are discussed, including the general maintenance of cellular energy levels and the more specific inhibition of molecules such as the nicotinamide adenine dinucleotide-dependent deacetylase, sirtuin 1 (SIRT1).
Keywords
Introduction
There is a growing body of evidence that diet and nutrition play a direct role in maintaining neuronal health. In particular, dietary factors can influence the onset and progression of Parkinson’s disease (PD), and potentially its amelioration.1,2 The emerging pattern from this body of research is that there are clear consequences to an imbalance in dietary factors on the production and maintenance of mature neurons.
Our research and that of others suggest that vitamins are essential both for the formation of neurons and their survival. Here, we review nicotinamide and associated active metabolites. We discuss nicotinamide’s role in the maintenance of mature central nervous system (CNS) neurons; its influence on neuronal health and survival during ageing, injury, and disease; and its potential as a therapeutic for neurodegenerative disease.
Vitamins and Their Role in Health
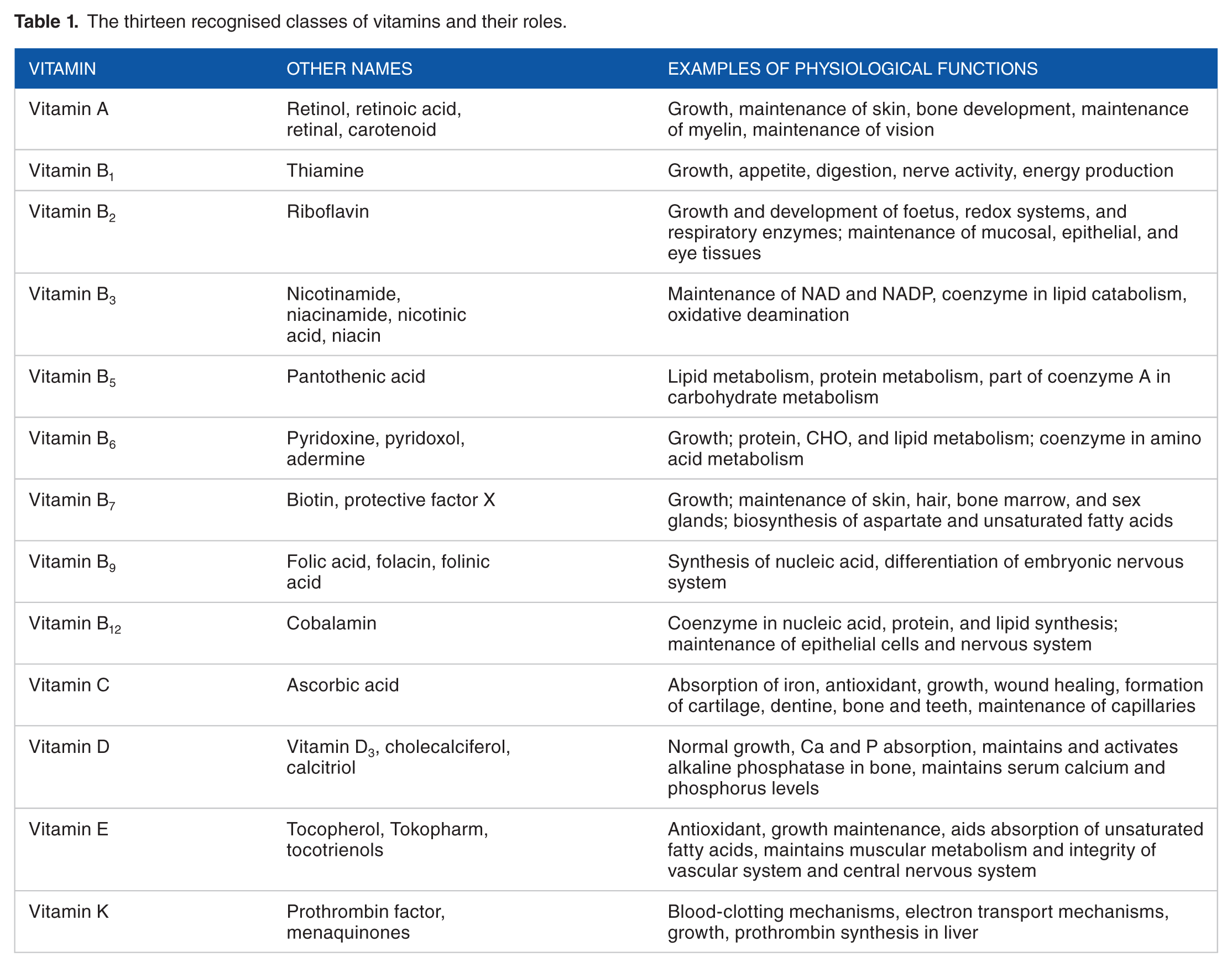
During the last century, a new class of nutritional supplements was identified. These ‘vitamins’ were defined as biologically active organic compounds essential for normal health and growth, which cannot, or can only partially, be synthesised by the human body. Grouped by their biological and chemical activity, 13 classes of vitamins (Table 1) are currently recognised, having diverse biochemical functions such as regulation of cell and tissue growth, mineral metabolism, acting as coenzymes in metabolism, and directing cell differentiation. 3 Thus, vitamins are essential for the development and maintenance of the body, with their deficiencies leading to conditions affecting multiple systems, such as pellagra, scurvy, rickets, bleeding disorders, and vulnerability to infections. 4 If untreated, vitamin deficiencies can lead to significant ill health and potentially death.
The thirteen recognised classes of vitamins and their roles.
Nicotinamide, Nicotinamide Adenine Dinucleotide, and Neuronal Health
Nicotinamide, the water-soluble amide form of vitamin B3, is a key component of the metabolic pathway involved in the production of nicotinamide adenine dinucleotide (NAD+). One source of nicotinamide is the diet, via intake of eggs, meat, fish, and mushrooms. A second source of nicotinamide is the metabolism of endogenous tryptophan, an essential amino acid. Nicotinamide can also be generated from niacin via the formation of NAD+.
Nicotinamide is stored in only small quantities in the liver, with most being either excreted or catabolised to provide other key metabolic products. It is difficult to achieve adverse effects from excessive intake, even with pharmacologically high doses, but overdose can cause hepatotoxicity in rare cases. 5
The enzyme, nicotinamide phosphoribosyltransferase (NAMPT), catalyses the synthesis of nicotinamide mononucleotide (NMN) from nicotinamide (Figure 1). Its role in the metabolic pathway for the biosynthesis of NAD (oxidised form NAD+; reduced form NADH) suggests its importance in cells that are sensitive to decreases in NAD levels, such as neurons. 6 NAD homeostasis has also been found to be altered with ageing7–10; thus, by influencing levels of NAD+ within neurons, nicotinamide may play a key role in neuronal maturation and neuroprotection.

Simplified schematic representation of the key pathways for the metabolism of nicotinamide, niacin, and tryptophan in the production of NAD+.
The enzyme NMN adenylyltransferase (NMNAT) converts NMN to NAD+ (Figure 1). Three isozymes, NMNAT1, 2, and 3, are localised to the nucleus, cytoplasm, or mitochondria, respectively. 11 An increase in NMNAT activity has been shown to lead to axonal protection in cultured neurons undergoing Wallerian degeneration, through a rise in nuclear NAD levels, leading to activation of the NAD-dependent protein deacetylase sirtuin 1 (SIRT1),12,13 implicating nicotinamide indirectly in neuroprotection.
In humans, nicotinamide undergoes some level of degradation, primarily through N-methylation to N-methyl nicotinamide via activity of the enzyme nicotinamide N-methyltransferase (NNMT). As mentioned above, the remaining metabolism of nicotinamide produces the NAD coenzymes in both the oxidised and reduced forms (NAD+ and NADH) in addition to nicotinamide adenine nucleotide phosphate, which is vital in mitochondrial respiration to produce adenosine triphosphate (ATP), as well as being implicated in more than 200 enzymatic reactions including those conferring cell protective and antioxidant roles (Figure 1).14–16
NAD+ can also be generated via tryptophan metabolism within the liver and kidneys 17 and from dietary nicotinic acid and niacin. Tryptophan can be metabolised into small amounts of nicotinic acid mononucleotide (NAMN) that can then be converted to NAD+. However, 60 mg of tryptophan is required to yield the equivalent amount of NAMN generated from 1 mg of niacin. 18 Therefore, tryptophan is not a necessary supplement to many Western, niacin-rich diets, 19 although tryptophan alone can be enough to prevent niacin deficiency. 17 Tryptophan metabolism is a 9-step process and the first part of this, known as the kynurenine pathway, 17 is altered in a number of neurodegenerative diseases including PD, Huntington’s disease (HD), and Alzheimer’s disease (AD)20,21 as well as other neurological disorders. 22 This disruption may increase the production of neurotoxins21–23 while also reducing NAD+ levels, leaving neurons more susceptible to damage. Thus, the finely balanced relationship between nicotinamide and NAD+ may greatly influence neuronal health.
Nicotinamide in the Peripheral Nervous System
Nicotinamide has been linked with Wallerian degeneration, ie, the axon degeneration that occurs distal to an injury or severance within axons of the peripheral nervous system. In peripheral nerve explants from a mouse mutant with slowed Wallerian degeneration (the Wlds mutation), Wlds acts to protect severed neurons, in conjunction with Nmnat1 and SirT1, with Nmnat overexpression also able to rescue degenerating axons.24,25 More recent studies suggest that NMNAT acts as a molecular chaperone to prevent protein misfolding and so protect key processes within neurons. 26 Another piece of evidence to suggest a role for nicotinamide in axon degeneration is the observation that NMN accumulates after nerve injury but prior to peripheral axon degeneration, and this can be ameliorated by inhibition of NAMPT, involved in the conversion of nicotinamide to NMN. 27
Nicotinamide has been proposed to be a key player in peripheral neuropathy within the eye. Overexpression of Nmnat1 protected retinal ganglion cells (neurons) from axonal degeneration and cell death after ischaemic insult and chronic elevation of intraocular pressure, a model for glaucoma. 28 In an aged mouse in vivo model, oral delivery of nicotinamide or enhanced expression of the Nmnat1 gene prevented both retinal ganglion cell soma loss and thinning of the retinal nerve fibre layer. 29 Similarly, in a mouse model of diabetes-induced neuropathy, sensory nerve endings within the cornea were protected through administration of nicotinamide riboside. 30 This effect could not be attributed to control of glucose alone, suggesting that other subcellular mechanisms are involved.
Another disease affecting the retinal pigment epithelium (RPE), and thus indirectly the photoreceptors transmitting sensory information to the optic nerve, is age-related macular degeneration (AMD). In a recent study, nicotinamide was shown to ameliorate the disease phenotype in RPE cells generated from pluripotent stem cell lines derived from patients with AMD, with nicotinamide and its associated pathways being proposed as targets for therapies for AMD. 31
Nicotinamide in the CNS
A number of studies indicate that nicotinamide is essential for the growth and maintenance of the CNS, acting to promote neuronal differentiation and neuronal survival, respectively. For example, nicotinamide appears to enhance and accelerate the conversion of embryonic stem cells to neural progenitors32,33 and neuronal differentiation from precursors 34 suggesting a key role in neural development.
There is a wealth of evidence to suggest that NAD+ metabolism has a direct influence on neuronal survival in the CNS.35,36 NAD+ is an important substrate that acts on 3 major classes of enzymes: the sirtuin family (SIRTs), the poly(ADP-ribose) polymerases (PARPs) and related adenosine diphosphate (ADP)-ribose transferases (ARTs), and the cyclic ADP-ribose (cADPR) synthases, CD38 and CD157. A by-product of SIRT, PARP, and ART activity is nicotinamide. Nicotinamide can inhibit the activity of these enzymes through binding to NAD+. In addition, neurons contain only low levels of the enzyme NAMPT, required for the first step in the conversion of nicotinamide to NAD+, potentially lowering its availability in these cells. NAD+ levels decrease with ageing 10 and this may be linked to lowered levels of NAMPT (for an excellent review see the work by Verdin 35 ). Further evidence to support this comes from studies where the class of aminopropyl carbazole chemicals P7C3 has been found to exert neuroprotective effects in models of PD, 37 stroke, 38 and amyotrophic lateral sclerosis 39 through activation of NAMPT. 40
There is evidence that nicotinamide can freely cross the blood-brain barrier in both directions. 41 Interestingly, one study has suggested that this transport is not affected in neurodegenerative disease, indicating that systemic nicotinamide could be given as a treatment without fear of reduced access to the CNS. 42 The NNMT messenger RNA (mRNA) is expressed in multiple CNS regions including the spinal cord, temporal lobe, medulla, cerebellum, and within the basal ganglia in the subthalamic nucleus, caudate nucleus, and the dopamine neurons of the substantia nigra, of particular relevance to PD. 43 These findings highlight the capacity of nicotinamide to influence neuronal differentiation and health, fuelling interest in potential applications as a neuroprotective agent.
The Role of Nicotinamide in Neuronal Injury, Ischaemia, and Stroke
Since the turn of the century, nicotinamide has been recognised as a key player in neuroprotection and neurorestoration in animal models of ischaemia.44,45 Nicotinamide has been shown to protect neuronal cells in a rodent model of ischaemic stroke and this effect is concentration dependent. In the early stages of developing cerebral infarction in the ischaemic brain, decreased levels of NAD+ are observed, preceding neuronal apoptosis. Studies have shown that intraperitoneal injection of 500 mg/kg nicotinamide up to 2 hours after ischaemia decreased the infarct volume of rats and improved both sensory and motor behaviour when compared with non-treated animals. 46
Prolonged hypoxia followed by re-oxygenation (reperfusion) of neural tissue leads to impairment of NAD+/NADH recycling, termed hyperoxidation. Pre-treatment with nicotinamide can improve neuronal function, reduce NADH levels, and restore ATP levels. 47 A similar effect was observed when the PARP-1 inhibitor PJ-34 was applied. Nicotinamide can inhibit PARP activity, consequently enhancing NAD production, and this may be one mechanism of neuroprotection (discussed later in this review). Niacin metabolism may lead to long-term restoration of the blood and oxygen supply to damaged neurons. Niacin given 24 hours after induction of experimental stroke in the rat significantly increased levels of high-density lipoprotein cholesterol. This in turn promoted angiogenesis, arteriogenesis, and local cerebral blood flow, reducing functional deficits. 44
Traumatic brain injury (TBI) is an area where nicotinamide may have a role as a therapeutic agent. Although the initial impact causing the trauma is highly damaging, the secondary sequelae create much of the lasting damage, through mechanisms such as inflammation, free radical generation, and excitotoxic cell death. Nicotinamide’s wide ranging influence on different cellular processes has made it a molecule worth exploring in TBI. Vonder Haar et al showed that infusion of nicotinamide via osmotic minipumps, starting 30 minutes following a controlled cortical impact injury, significantly reduced the lesion size. This neuroprotection was correlated with improvement of sensory, motor, and cognitive skills, with animals showing improved scores on the bilateral tactile adhesive removal task, locomotor placing task, and reference memory paradigm of the Morris water maze, respectively. 48 A more recent study showed further improvements in reducing cortical neuron loss after a contusion injury when nicotinamide was co-administered with progesterone. Results showed a significant decrease in cavitation, degenerating neurons and reactive astrocytes. Transcriptional profiling suggested a reduction in genes in both inflammatory and immune pathways. Progesterone and nicotinamide–co-treated animals reached higher scores on adhesive removal and forelimb placing tasks, compared with groups that received either treatment alone. 49
The Role of Nicotinamide in Neurodegenerative Disease
Alzheimer’s disease
Alzheimer’s disease is one of the most common neurodegenerative diseases, affecting perhaps 30 million people worldwide, 50 who suffer slow cognitive decline. The characteristic pathology of AD involves the presence of amyloid β (Aβ) plaques and neurofibrillary tangles. 51 The exact cause of AD is unknown, although genetic, environmental, and developmental factors are thought to be involved. 52 Cases are highest in developed countries and are expected to rise, with the biggest increase occurring in developing countries. 53 There is an urgent need to identify neuroprotective, and ideally neuro-regenerative, treatments for AD, but clinical trials have thus far failed to reliably demonstrate efficacy in a substantial number of patients. However, several lines of evidence suggest that nicotinamide or related molecules may offer therapeutic benefits for patients with AD.54–61
For example, severe tryptophan/niacin deficiency leads to the syndrome pellagra, where patients can develop neurological deficits manifesting as dementia, and described as ‘premature ageing’. 62 Symptoms are similar to AD and include psychosis, disorientation, memory loss, and confusion, which can all be combated by niacin supplementation. 63 Although pellagra is seen primarily in young people in areas where diets are based mainly on corn (eg, Africa and India), it also occurs in adulthood in Western societies, eg, in alcoholics who are usually deficient in numerous vitamins or as a consequence of eating disorders such as anorexia nervosa. 14 Niacin deficiency in ageing populations has been linked with dementia. In a study between 1993 and 2002 in the Chicago community, dietary levels of niacin were shown to be inversely related to the onset of AD, measured through at least 2 clinical cognitive evaluations. 64 It is not clear from these data whether niacin or nicotinamide may be the more important metabolite.
Nicotinamide and niacin produce cellular and molecular effects that may be relevant to AD. Elevated total cholesterol and low-density lipoproteins are linked directly to the pathology of AD. 44 Cholesterol in neurons contributes to Aβ formation and accumulation, and it has been suggested that increased levels of membrane cholesterol can make hippocampal neurons more sensitive to insults such as tau toxicity. 65 Niacin decreases cholesterol levels both in the serum and intracellularly, which may offer protection in AD. Niacin upregulates peroxisome proliferator–activated receptor γ (PPARG) mRNA expression, promoting cholesterol efflux and so reducing cellular levels. Niacin also has been shown to upregulate liver X receptors, stimulation of which facilitates clearance of Aβ42, 44 and may improve memory in an AD mouse model. 66
Within the neuron, NAD+ serves as a substrate for the synthesis of cADPR, used in calcium signalling, important for synaptic plasticity. This is particularly important in the hippocampus, a structure critical for learning and memory. Thus, through maintaining levels of NAD+, nicotinamide could protect against age-dependent neuronal degeneration in the hippocampus. Interestingly, however, Young and Kirkland showed that decreased niacin intake and cADPR levels actually led to enhanced ability for spatial learning in adult male rats. When the diet was supplemented with nicotinamide, spatial learning ability then decreased. 67 This suggests that nicotinamide’s relationship with hippocampal neurons and learning and memory may be more complex than predicted. Interestingly, the enzyme Nmnat2, which is involved in the conversion of nicotinamide to NAD+, has been linked to neuroprotection against tauopathy in a mouse model of dementia. Nmnat2 transcription was seen to be downregulated prior to neurodegeneration in a transgenic mouse model possessing a mutation associated with frontotemporal dementia. Injection of adeno-associated viruses overexpressing Nmnat2 in the hippocampus of these mice from 6 weeks of age reduced the extent of neurodegeneration observed at 5 months. 68 Lower levels of Nmnat2 mRNA and protein have also been observed in patients with AD, and its activity is linked to tau clearance. 69 Green et al 55 report that oral nicotinamide selectively reduces phosphoThr231-tau in a mouse model of AD, through a mechanism similar to SirT1 inhibition. This increased levels of microtubule stability–associated proteins and reduced cognitive deficits but did not affect Aβ pathology.
Mitochondrial dysfunction and bioenergetic deficits interrupt synaptic plasticity and impair learning and memory. These mechanisms are increasingly proposed to be key to AD. 56 Neuronal mitochondrial function can be improved through increased NAD+ and the activity of SIRT1 and SIRT3. 56 This has been achieved in a mouse model of AD, where nicotinamide treatment diminished learning and memory impairment. 59 Nicotinamide has also been shown to reduce oxidative stress in ex vivo and in vivo rat models of AD.61,70
The evidence outlined above has underpinned several clinical trials for AD, using nicotinamide or NADH. In 1996, NADH was reported to improve mini mental state examination scores in patients with AD, although this was an open-label trial for 8 to 12 weeks, with only 17 subjects and no controls. 71 Rainer et al 72 failed to detect improved cognitive effects of NADH in patients with dementia (including AD). In 2004, a randomised double-blind clinical trial using NADH with patients with AD reported a halt in cognitive decline and superior verbal fluency (compared with placebo; n = 12 in treatment group). 73 A 2017 nicotinamide clinical trial (Safety Study of Nicotinamide to Treat Alzheimer’s Disease; NCT00580931) reported no increase in adverse events, supporting the relative safety of high (1500 mg, twice daily) nicotinamide doses. 74 No improvements in the monitored cognitive functions were detected as the number of patients was small (n = 15) and the time-course was relatively short (24 weeks). 74 A further clinical trial is investigating the effects of nicotinamide on the phosphorylation of tau (Nicotinamide as an Early Alzheimer’s Disease Treatment [NEAT]; NCT03061474) and is due to be completed in February 2019.
Parkinson’s disease
One particular neurodegenerative disorder that may be influenced by diet and nutrition is PD. A significant hallmark of this disorder is the death of midbrain dopamine neurons within the substantia nigra, leading to an imbalance in activity within the basal ganglia circuitry deep within the brain, manifesting in reduced movement (akinesia), rigidity, and tremor. About 95% of cases of PD cannot currently be attributed to genetic defects; therefore, science research has focussed on environmental factors that may influence the health of mature substantia nigra dopamine neurons.
Parkinson’s disease is characterised by neuronal inclusions comprising α-synuclein aggregates. Although the cause of the disease is currently unknown, one hypothesis is that the dopamine neurons are compromised through oxidative stress, and more recently, it has been suggested that this oxidative stress may originate in the gastro-intestinal tract, leading to neuronal damage. People with PD have been shown to have increased intestinal permeability as well as α-synuclein aggregates and higher levels of oxidative stress in the gastro-intestinal tract. Although there is only limited evidence for a direct link between diet and PD, these data suggest that diets high in saturated fat from animal sources may have a negative impact on neuronal health, whereas unsaturated fats and foods containing antioxidants may be protective, by reducing inflammation and oxidation. 75
Vitamin B3 intake has been suggested to play both protective and detrimental roles in PD. 14 NAD+ levels are found to be decreased in patients with PD 76 and a reduced risk of PD is associated with higher consumption of foods containing niacin.77,78 The patients with PD taking niacin supplementation for other disorders reported an easing of the symptoms, although doses were stopped due to adverse side effects.79,80
NADH is fundamental for the normal functioning of mitochondrial complex 1, which is established to be defective in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism 81 and idiopathic PD.82,83 NADH is also integral to the production of tetrahydrobiopterin, 84 a co-factor necessary for tyrosine hydroxylase, the rate-limiting enzyme in catecholamine biosynthesis, also deficient in PD. In addition, NADH is linked to reduced glutathione, an important antioxidant shown to be insufficient at the early stages of PD.15,85 Thus, the ability for nicotinamide to increase levels of NADH or ATP might be therapeutic for compromised midbrain dopamine neurons. In 2 mouse models of PD, nicotinamide has demonstrated neuroprotective properties by attenuating striatal dopamine depletion and protecting substantia nigra pars compacta neurons in acute MPTP-treated mice. 16
Levels of nicotinamide- NNMT have been shown to be increased in the cerebrospinal fluid and within specific neuron populations including the midbrain dopamine neurons, of patients with PD, suggesting a role in pathogenesis.43,86 Higher NNMT levels have been proposed to cause an increase in conversion of nicotinamide to N-methyl nicotinamide, structurally related to the toxin N-methyl-4 phenylpyridinium (MPP+), the active MPTP derivative that selectively destroys dopamine neurons. 85 In addition, increased NNMT activity leads to lowered mitochondrial complex 1 activity and impaired mitochondrial function, resulting in neurodegeneration.15,87 Given this, a high level of nicotinamide obtained from meat-rich diets has been proposed as a nutritional factor that, in excess, may predispose dopamine neurons to mitochondrial stress, triggering neuronal apoptosis and leading to PD.62,88 In support of this theory, our own work has shown that cultured stem cell–derived neurons respond positively to supplementation with nicotinamide within a dose range of 5 to 10 mM in vitro, but that a 20-mM dose is highly toxic to all neurons. 32
Altering the course of PD through dietary means is difficult, and further experimentation is needed to determine whether this modulation is impactful; however, potential exists for supplements such as prebiotics to beneficially modify the gut milieu, reducing constipation in individuals with PD. Due to the potential role of the gastrointestinal barrier in exposure to injurious factors, therapeutic intervention through whole foods, dietary patterns, and supplemental nutrition (probiotics, prebiotics, and synbiotics) may positively impact intestinal milieu and result in reduced inflammation and oxidation and reduced risk for PD.
Huntington’s disease
Similar to PD, neurodegeneration in HD has been associated with impaired mitochondrial function. Loss of GABAergic medium spiny projection neurons in the striatum is thought to occur through the mutant form of the protein huntingtin interfering with normal cellular processes such as respiration and energy metabolism, leading to neuronal dysfunction. Potential downstream effects on the neurons include altered protein trafficking and synaptic transmission, excitotoxicity through overstimulation of excitatory glutamatergic receptors, altered calcium levels, generation of free radicals, and neuronal apoptosis. For over 2 decades, it has been known that mitochondrial toxins such as malonate or 3-nitropropionic acid (3-NP) administered to rodents can induce striatal neuron degeneration.89–91 Administration of nicotinamide can attenuate the extent of striatal lesions produced by malonate and this effect has been seen both with a continual release paradigm (via infusion pump over 7 days) or when nicotinamide was given intraperitoneally just prior to and following the lesion surgery. 92 There was an additive effect on neuroprotection when nicotinamide was delivered in combination with coenzyme Q10, an essential component of the electron transport chain and a free radical scavenger, suggesting that multiple pathways may be involved in the neuroprotection observed. 92
Metabolic toxins such as malonate provide a crude insult to medium spiny striatal neurons. More recent research has assessed the effect of nicotinamide in a transgenic model of HD (the B6.HDR6/1 mouse model that expresses a mutant form of the human HD gene where exon 1 contains an expanded stretch of approximately 125 CAG repeats), thought to more closely replicate the disease process. Nicotinamide delivered either through an osmotic minipump or in the drinking water increased brain levels of brain-derived neurotrophic factor (BDNF) and PPARG (PGC-1α) concomitantly improving movement control, measured in the open-field, rotarod, and balance beam tasks. Interestingly, behavioural improvement was not associated with any decrease in abnormal aggregation of Huntington protein and did not prevent late-stage weight loss. 93 Conversely, nicotinamide administered to an alternate transgenic model (YAC128 mice, expressing the entire human HD gene with exon 1 containing 128 CAG repeats) did not lead to an improvement in motor behaviours and in wild-type control animals actually worsened their motor performance. 94 The latter study compared nicotinamide directly against resveratrol to investigate their effects as either a SirT1 inhibitor or activator, respectively. In contrast to nicotinamide, resveratrol improved motor performance. Interestingly, both molecules had a positive effect on cultured striatal and cortical neurons derived from YAC128 mice, suggesting that opposed actions on SirT1 could lead to a similarly positive survival outcome. 94
Mechanisms for neuroprotection by nicotinamide
Rather than just being a nutritional factor, nicotinamide has been shown to both prevent and reverse the injury of neuronal and endothelial cells. Nicotinamide can support DNA stability and can maintain membrane integrity, preventing cellular injury, phagocytosis, apoptosis, and vascular clot formation. 45
One mechanism by which nicotinamide may act is simply to restore ATP levels within neurons. For instance, MPTP, used to create a model of PD, inhibits the mitochondrial complex 1 selectively within dopaminergic neurons, leading to ATP depletion and formation of reactive oxygen species (ROS), ultimately leading to cell death. Anderson et al 16 propose that sub-acute MPTP exposure leading to an energy crisis in the neurons can be ameliorated by nicotinamide, through restoration of intracellular NAD+ and ATP levels.
Nicotinamide’s neuroprotective capacity has been linked to PARP, an enzyme implicated in DNA repair and cell death that, in excess, causes depletion of both NAD+ and ATP. PARP is activated in response to DNA damage, possibly through oxygen-free radicals, where it catalyses NAD+ into ADP-ribose and nicotinamide. 95 Overactivation of PARP leads to NAD+ depletion and lowers ATP levels. 62 Nicotinamide can inhibit the activity of PARP, thereby protecting cells from oxidative stress, apoptosis, and necrotic forms of cell death.
The importance of nicotinamide in replenishment of NAD+ levels also links with the transmembrane glycoprotein CD38. Through consumption of NAD+, CD38 synthesises and hydrolyses cADPR and ADP-ribose 96 and may have a role in regulating NAD levels. 97 The activity of this NADase has been found to increase with age and is responsible for age-related decline of NAD. 98 Cells overexpressing CD38 not only have lower NAD levels but also have a reduction in proteins associated with antioxidant defence, leaving them more susceptible to oxidative stress. 99 CD38 is also implicated in the degradation of NMN 98 meaning that an increase in activity could not only degrade NAD but also lower production levels, necessitating the need for extra nicotinamide. Another protein responsible for NAD depletion is sterile alpha and TIR motif containing 1 (SARM1). After axonal damage expression of SARM1 mediates axon degeneration while depleting NAD+, however, degeneration can be blocked by expression of NMNAT1 and Nampt.100,101
An alternative mechanism of action for nicotinamide is through inhibition of the NAD-dependent deacetylase SIRT1. SIRT1 is expressed throughout the CNS, predominantly within the nucleus of neurons, and is found within brain regions that are susceptible to neurodegeneration seen with ageing. 102 It has been associated with the conversion of neural progenitors to a neuronal fate 103 and then to specific neuronal phenotypes, including motor neurons of the spinal cord. 104 Changes in SIRT1 levels have also been implicated in neuronal ageing and neurodegeneration.12,105 Increased SIRT1 levels have been associated with neuroprotection in AD through targeting both Aβ and tau proteins. In PD, SIRT1 may protect dopamine neurons through activating heat shock factor 1 (HSF1) levels in dopamine neurons, and in HD, through activation of CREB-regulated transcription coactivator 1 (TORC1) and subsequently BDNF levels. 105 However, experimental data have to be interpreted in the context of NAD levels within neuronal populations as sirtuins require NAD for their bioactivity and therefore SIRT1 may show opposite effects when present with either low or high levels of NAD, respectively.
Nicotinamide has also been shown to influence DNA degradation via a number of cell pathways, eg, activating protein kinase B (Akt), which phosphorylates the forkhead transcription factor (FKHRL1), inhibiting apoptosis. Activation of Akt maintains mitochondrial membrane polarisation, preventing the release of cytochrome C, thus averting cellular injury. 45 Nicotinamide can also act as an inhibitor of caspase 1, caspase 3, and caspase 8 during cellular injury, in turn preventing the release of cytochrome C and inhibiting apoptosis.45,106
Nicotinamide may also act to prevent neurodegeneration through altering calcium signalling. Calcium signalling plays a major role in many neuronal processes including axon elongation and response to external stimuli. However, there is evidence from studies on Wallerian degeneration and neurodegenerative disease that axonal degeneration leads to an inability to control calcium levels, and that a rise in calcium within the axon creates neurotoxicity, causing neuronal death. 107 Aberrant calcium signalling has also been implicated in the mechanism by which neuroinflammation causes neurodegeneration, suggesting that calcium receptors may be a target for neuroprotective therapies. 108 Nicotinamide has been shown to be an inhibitor of cADPR in sea urchin eggs by interfering with the mobilising activity of the key intracellular signalling molecules: β-NAD+, cyclic GMP, and nitric oxide – modulators of cADPR synthesis. 109
Nicotinamide and NAD also have implications in immune cell modulation 110 and with activated microglia found at sites of neurodegeneration, 111 these molecules could work through anti-inflammatory means. GPR109A, an anti-inflammatory G-protein receptor present on macrophages, has been found in higher levels in patients with PD 76 ; however, treatment with niacin reduces these expression levels. 80
More evidence for the mechanisms by which nicotinamide confers neuroprotection comes from studies of the vascular system. A body of evidence suggests that vascular ageing and associated endothelial breakdown are linked to an increase in ROS within vascular cells such as the progenitors that are required for endothelial repair. 112 Lowering levels of ROS, eg, through dietary caloric restriction may stimulate ROS-dependent protective pathways, such as those involving SIRT1, and have anti-inflammatory effects on endothelial cells. 113 Nicotinamide itself has been investigated in mouse models as a treatment option for pre-eclampsia. Increased dietary intake of nicotinamide was shown to improve the health of both mothers and pups, decreasing blood pressure and endotheliosis. The authors suggest that the mode of action of nicotinamide was through restoration of foetal ATP synthesis, mostly likely through inhibition of cADPR. 114
Clearly, there are a number of mechanisms implicated for nicotinamide’s role in neurodegeneration or neuroprotection, due to its activity in so many cellular processes conferring energy generation and cellular protection and repair, and evidenced from numerous body systems as well as the CNS. Teasing out specific downstream effects of nicotinamide’s activity remains a challenge, but one worth exploring.
Summary
There is a growing body of evidence that nicotinamide is implicated in neuronal differentiation and health, neuronal injury, and neurodegeneration in the CNS. Changes in nicotinamide levels have been linked with AD, PD, and HD, and nicotinamide treatment in animal models has shown amelioration of neurodegeneration and associated behavioural recovery. Equally, there is evidence of nicotinamide being used as a restorative agent in animal models of neuronal injury and ischaemia.
The plethora of intracellular systems influenced by nicotinamide levels makes it difficult to determine precise mechanisms of action by this dietary metabolite. However, it is becoming clear that nicotinamide should be titrated to balanced levels in the CNS to avoid neural sequelae caused by either too little or too much nicotinamide within mature neurons.
With this in mind, supporting neuronal health through good dietary supplementation and management of small bioactive molecules such as nicotinamide appears an exciting and achievable prospect.
Footnotes
Acknowledgements
This concise review article was specifically commissioned for the special issue of the International Journal of Tryptophan Research on tryptophan metabolism and neurodegenerative diseases. We summarise global research into the mechanisms by which the active tryptophan metabolite nicotinamide may cause or prevent neurodegenerative disease, with specific focus on the CNS. Thus, this article aims to help readers to identify high-quality evidence to develop an informed opinion on the potential of nicotinamide to prevent or combat neuronal death.
Funding:
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
RAF wrote the first draft of the manuscript. SMG, ELG, and SIJ contributed to the writing of the manuscript and agree with manuscript results and conclusions. ELG and SIJ made critical revisions and approved final version. All authors reviewed and approved the final manuscript.
Disclosures and Ethics
As a requirement of publication, authors have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. The external blind peer reviewers report no conflicts of interest.