
Abstract
Background
Breast cancers represent a heterogeneous group of diseases; approximately 7% may be attributed to inherited pathogenic variants in BRCA1 and BRCA2, with exon 11 of BRCA1 representing the most frequently mutated region globally.
Objectives
This study aimed to investigate the frequency and nature of sequence variants in BRCA1 exons 11 and 20 and BRCA2 exon 11 in a Sudanese cohort, and to determine whether these established mutational hotspots harbor recurrent pathogenic variants in this underrepresented population.
Design
This was a case-control study conducted at Shendi’s Tumor Treatment and Cancer Research Center in Northern Sudan.
Methods
The study included fifty-two female breast cancer patients and thirty healthy female controls aged at least 18 years. Demographic data and blood samples were collected for genomic DNA extraction. Polymerase Chain Reaction (PCR) and Sanger sequencing were performed for BRCA1 (exons 11 and 20) and BRCA2 (exon 11). Variants were classified using ACMG/AMP criteria and analyzed using bioinformatics tools and SPSS.
Results
Invasive ductal carcinoma was the predominant histological type, significantly associated with grade II tumors (P = 0.0001). Non-hereditary breast cancers were more prevalent (55.8%), with second-degree relatives most commonly affected in hereditary cases (69.6%). Three BRCA1 sequence variants were identified, all classified as benign or likely benign. These variants were found at comparable frequencies in cases (13/52, 25.0%) and controls (8/30, 26.7%; P = 0.863). Variant presence was significantly associated with Jaalia ethnicity (P = 0.047) and observed exclusively in IDC cases, though these associations did not reach statistical significance for tumor characteristics.
Conclusion
No pathogenic variants were identified in BRCA1 exons 11 and 20 or BRCA2 exon 11 in this Sudanese cohort. Given that BRCA1 exon 11 constitutes approximately 60% of the coding sequence and harbors the majority of pathogenic variants in other African populations, these findings suggest that mutational hotspots may differ in this population. Expanded genomic studies encompassing complete coding regions are warranted.
1. Introduction
Breast cancer represents the most prevalent malignancy affecting women globally. According to the International Agency for Research on Cancer (IARC) Globocan 2022, approximately 2.26 million new cases were diagnosed, and 670,000 deaths were reported globally. 1 Breast cancers constitute a heterogeneous group of diseases characterized by genetic and epigenetic alterations that disrupt normal cellular regulatory mechanisms, leading to uncontrolled proliferation, evasion of apoptosis, angiogenesis, and metastasis. 2 Epidemiological studies indicate that approximately one in eight women will develop breast cancer during their lifetime. 1 The disease is influenced by a complex interplay of genetic, hormonal, and environmental factors, with sequence variants in the BRCA1 (Breast Cancer Gene 1) and BRCA2 (Breast Cancer Gene 2) being among the most significant hereditary risk factors. 2 While the majority of breast cancer cases arise sporadically, approximately 7% may be attributed to germline sequence variants in predisposing genes, which confer a markedly increased lifetime risk. Given the molecular heterogeneity of breast cancers, precision oncology approaches tailored to distinct molecular subtypes are essential for optimizing therapeutic outcomes. 3
BRCA1 sequence variants are widely recognized for their critical role in breast cancer predisposition, primarily due to their function in DNA (Deoxyribonucleic Acid) double-strand break repair via homologous recombination (HR) pathways. 4 Deficiency in homologous recombination repair (HRD) is not only a hallmark of ovarian cancer but is also increasingly implicated in breast cancer pathogenesis, influencing both prognosis and treatment response. 4 The presence of BRCA1 sequence variants in breast cancer patients is associated with an elevated risk of tumor development, more aggressive disease phenotypes, and reduced responsiveness to conventional therapies. 1 Notably, BRCA1 exon 11 is the largest exon, comprising approximately 60% of the coding sequence, and represents the most frequent site of pathogenic variants across diverse global populations, including North and West African cohorts.1,5 Understanding the mechanistic implications of BRCA1 sequence variants is vital for advancing molecular diagnostics, risk assessment, and the development of targeted therapeutic strategies, such as PARP (Poly(ADP-ribose) Polymerase inhibitors, which exploit synthetic lethality in HR-deficient tumors.2,3
In Africa, breast cancer incidence is rising. According to IARC Globocan 2022, approximately 188,000 new cases and 81,000 deaths were reported.1,5 Limited healthcare infrastructure, delayed diagnosis, and restricted access to advanced treatments contribute to disproportionately higher mortality rates compared to high-income countries. In Sudan, breast cancers represent a major public health challenge, accounting for approximately 34% of all female malignancies. 6 The annual incidence rate stands at approximately 25 cases per 100,000 women, equating to an estimated 3,700 new cases each year. 7 This increasing trend is driven by factors such as rapid urbanization, changes in reproductive patterns, and limited access to early screening programs. Consequently, the mortality rate remains alarmingly high, with breast cancers responsible for approximately 23% of cancer-related deaths among Sudanese women.8,9 These statistics underscore the urgent need for strengthening early detection programs, enhancing public awareness, and expanding access to modern therapeutic interventions.
This study aims to characterize sequence variants in BRCA1 exons 11 and 20 and BRCA2 exon 11 in Sudanese breast cancer patients, and to determine whether these regions—which are established mutational hotspots in other populations—harbor recurrent pathogenic variants in this underrepresented African cohort. Understanding population-specific genetic profiles is critical for improving genetic counseling, risk stratification, and precision medicine approaches in underrepresented African populations. By addressing these unique population-specific genetic variations, this study enhances the global understanding of breast cancer predisposition and reinforces the importance of integrating genetic screening into breast cancer management strategies, particularly in underrepresented populations.
2. Materials and Methods
2.1. Study Design, Aims, and Population
This case-control study evaluated BRCA1 and BRCA2 sequence variants in breast cancer patients to determine their prevalence and clinical significance in a Sudanese population. The objectives were to characterize genetic variants in selected exons, assess their associations with clinicopathological features, and provide data to support precision medicine approaches in underrepresented populations (Figure S1). The study included 52 women with breast cancer recruited from the Tumor Treatment and Cancer Research Centre in Shendi, Northern Sudan, and 30 healthy women with no history of cancer as controls. A STROBE checklist from the EQUATOR Network is provided in Supplemental Table S1 to ensure transparent reporting of observational data. 10
2.2. Inclusion and Exclusion Criteria
The study included female breast cancer patients aged ≥18 years from the Tumor Treatment & Cancer Research Centre who consented to participate. Healthy females aged ≥18 years without a cancer history served as control subjects. All participants provided informed consent before inclusion. Exclusion criteria encompassed individuals with previous cancer diagnoses, genetic disorders, or health conditions that could influence the results, as well as those unable or unwilling to give informed consent.
2.3. Sample Size Calculation
Sample size was calculated using the formula for proportion estimation: n = (Z2pq)/d2. Assuming an expected BRCA1/BRCA2 variant frequency of 7% (p=0.07) based on global averages, with a 95% confidence level (Z=1.96) and a 7% margin of error (d=0.07), the minimum required sample size was 51, rounded to 52 cases. The control group comprised 30 healthy women enrolled during the same period, yielding a case-to-control ratio of approximately 1.7:1.
2.4. Laboratory Procedures
2.4.1. Sample Collection and DNA Extraction
Genomic DNA was extracted from whole blood samples of both breast cancer patients and healthy controls using the iNTRON Biotechnology DNA (Deoxyribonucleic Acid) extraction kit (South Korea), in strict accordance with the manufacturer’s instructions.
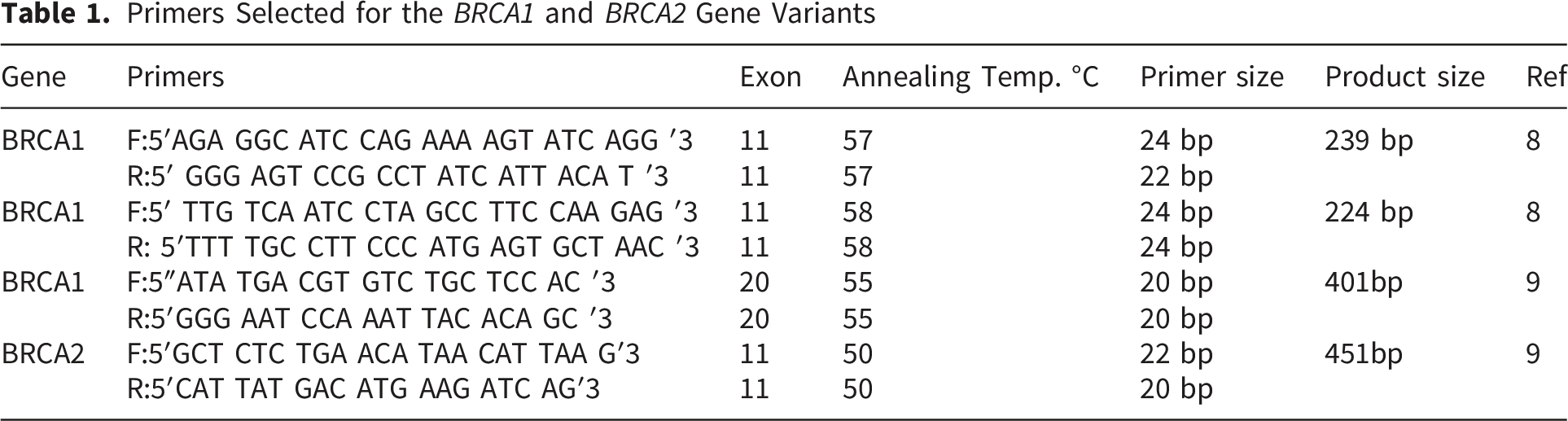
2.4.2. Primer Selection and DNA Target Amplification
Primers Selected for the BRCA1 and BRCA2 Gene Variants
2.4.3. DNA Amplification Procedure and Agarose Gel-Electrophoresis
The extracted and eluted DNA samples were subsequently amplified via polymerase chain reaction (PCR) targeting the specific genetic regions, utilizing the Maxime PCR PreMix Kit i-Taq 20 μl (iNTRON Biotechnology, South Korea), a ready-to-use master mix containing Taq DNA polymerase, dNTPs, and reaction buffer. For each sample, the reaction mixture was prepared by adding 15-17 μl of distilled water, 1-3 μl of template DNA, and 1 μl each of the forward and reverse primers to the pre-mixed components. The amplification was performed in a thermal cycler under the following conditions: an initial denaturation at 96°C for 5 minutes, followed by 35 cycles consisting of denaturation at 96°C for 30 seconds, primer annealing for 30 seconds at temperatures optimized for each primer set (50°C, 55°C, 57°C, or 58°C), and an elongation step at 72°C for 1 minute, with a final extension at 72°C for 10 minutes. Following amplification, the PCR products were resolved by electrophoresis on a 2% agarose gel stained with ethidium bromide. The gel was prepared by dissolving 0.5 g of agarose in 25 ml of 1X TBE buffer through heating in a microwave, cooling the solution to room temperature, and adding 2 µl of ethidium bromide before pouring it into a casting tray with a comb and allowing it to solidify for 30 minutes. For analysis, 10 µl of each PCR product and a 100 bp DNA ladder were loaded into the wells, and electrophoresis was conducted at 100V and 60 mA for 30-45 minutes. The resulting DNA bands were visualized and documented using a gel documentation system (Gel Mega) equipped with a digital camera and imaging software.
2.4.4. Sanger Sequencing and Bioinformatics Analysis
Sanger sequencing was performed by trained laboratory personnel using an ABI (Applied Biosystems) DNA Analyzer. PCR (Polymerase Chain Reaction) products were purified using ExoSAP-IT (Affymetrix, USA) to remove unincorporated dNTPs and primers. Sequencing reactions were prepared using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), with cycling conditions: 96°C for 1 minute, followed by 25 cycles of 96°C for 10 seconds, 50°C for 5 seconds, and 60°C for 4 minutes. Sequencing was performed in both forward and reverse directions to ensure sequence accuracy and confirm variant identification. The sequence chromatograms were analyzed using Finch TV 1.4.0 software. To assess the similarity of nucleotide sequences for BRCA1 and BRCA2, nucleotide BLAST (Basic Local Alignment Search Tool) and the NCBI (National Center for Biotechnology Information) database were utilized, referencing transcript variant 1 and protein accession numbers. 11 Any detected sequence variants were further evaluated through multiple sequence alignments using BioEdit software, and amino acid sequences were derived using the Expasy translation tool, followed by re-analysis with BioEdit version 7.0.9.1. 12
2.4.4.1. SNP (Single Nucleotide Polymorphism) Detection and Prediction
The single nucleotide polymorphism (SNP) data utilized for the computational analyses in this study correspond to the human BRCA1 gene, specifically referencing SNP ID MIM:113705, with RefSeq Gene accession NG_005905 located on chromosome 17; the analyses employed mRNA transcript variant 1 (accession number NM_007294, 7224 bp) and its corresponding protein isoform 1 (accession number NP_009225, 1863 amino acids), all of which were retrieved from the National Center for Biotechnology Information (NCBI) dbSNP database (https://www.ncbi.nlm.nih.gov/). To predict the functional consequences of the identified SNPs, a suite of four established in silico prediction tools was employed: SIFT (https://sift.bii.astar.edu.sg/) was used to assess the impact of amino acid substitutions on protein function based on sequence homology and physical properties; Polymorphism Phenotyping v2 (PolyPhen-2, https://genetics.bwh.harvard.edu/pph2/) was utilized to predict the possible impact of an amino acid substitution on the structure and function of a human protein using physical and comparative considerations; I-Mutant Suite (https://gpcr2.biocomp.unibo.it/cgi/predictors/IMutan3.0/IMutant3.0.cgi) was employed for the prediction of protein stability changes upon single point mutations; and Predictor of human Deleterious Single Nucleotide Polymorphisms (PhD-SNP, https://snps.biofold.org/phd-snp/phd-snp.html) was used to predict whether a given SNP is disease-related or neutral. Furthermore, tertiary protein structure modeling and detailed mutation analysis at the atomic level were conducted using the Project HOPE online software (https://www.cmbi.ru.nl/hope/input), which provides insights into the structural effects of the mutations by analyzing the altered physicochemical properties of the mutant amino acid residues.13-16
2.4.5. Data Collection and Statistical Analysis
Demographic and clinical data were collected through self-administered questionnaires and laboratory tests. Occupational status was categorized as “worker” (manual labor) or “employee” (salaried positions). Physical activity levels were self-reported by participants, and body mass index (BMI) was calculated using the standard formula weight (kg)/height (m2) and categorized according to World Health Organization guidelines. Classification of hereditary versus sporadic breast cancer was based on family history: hereditary cases were defined as patients with a documented first-, second-, or third-degree relative with breast cancer, whereas sporadic cases had no documented family history of the disease. Data on prior radiation exposure, which refers specifically to diagnostic or therapeutic medical radiation and excludes occupational exposure, were also collected. Statistical analyses were performed using IBM SPSS Statistics version 20. Descriptive statistics, frequency distributions, and Chi-square tests were used to analyze categorical variables.
3. Results
3.1. Clinicopathological and Demographic Characteristics of the Study Cohorts
The clinicopathological and demographic profiles of the breast cancer patient cohort are detailed in Supplemental Tables S2 through S6. An analysis of histological subtypes in relation to patient age (Table S2) revealed invasive ductal carcinoma (IDC) as the predominant form, accounting for 46 of the 52 cases (88.5%). Within the IDC subgroup, the highest frequency was observed in patients aged 51–60 years (n=21, 45.7% of IDC cases), followed by those aged 41–50 years (n=10, 21.7%). Other less frequent histological types included ductal carcinoma in situ (DCIS; n=2, 3.8%), lobular carcinoma (n=2, 3.8%), and Paget disease (n=2, 3.8%). The correlation between histological type and tumor grade is presented in Table S3. A significant association was observed for IDC, with the majority of cases classified as grade II (n=31, 67.4%) and grade III (n=14, 30.5%), while only a single IDC case (2.1%) presented as grade I. This distribution was highly significant (p = 0.001), confirming a strong predilection for higher-grade tumors within the IDC subtype.
Regarding genetic predisposition, the cohort was classified based on hereditary status (Table S4). A total of 23 patients (44.2%) were identified as having a hereditary form of breast cancer, while the remaining 29 patients (55.8%) were classified as non-hereditary cases. Among those with hereditary cancer, the degree of affected relatives was second-degree (n=16, 69.6%), followed by first-degree (n=4, 17.4%) and third-degree (n=3, 13.0%) relatives (Table S5). An analysis of cancer types affecting the family members of these hereditary cases (Table S6) demonstrated that breast cancer was the most prevalent m/alignancy, occurring in 17 families (73.9%), while other, non-specified cancer types accounted for the remaining 6 cases (26.1%).
The Distribution of Sociodemographic Characteristics and Breast Cancer Risk Factors in the Study Population of Cancer Patients

3.2. Distribution of BRCA1 Sequence Variants in Cases and Controls
Detailed Characterization of Identified BRCA1 Sequence Variants
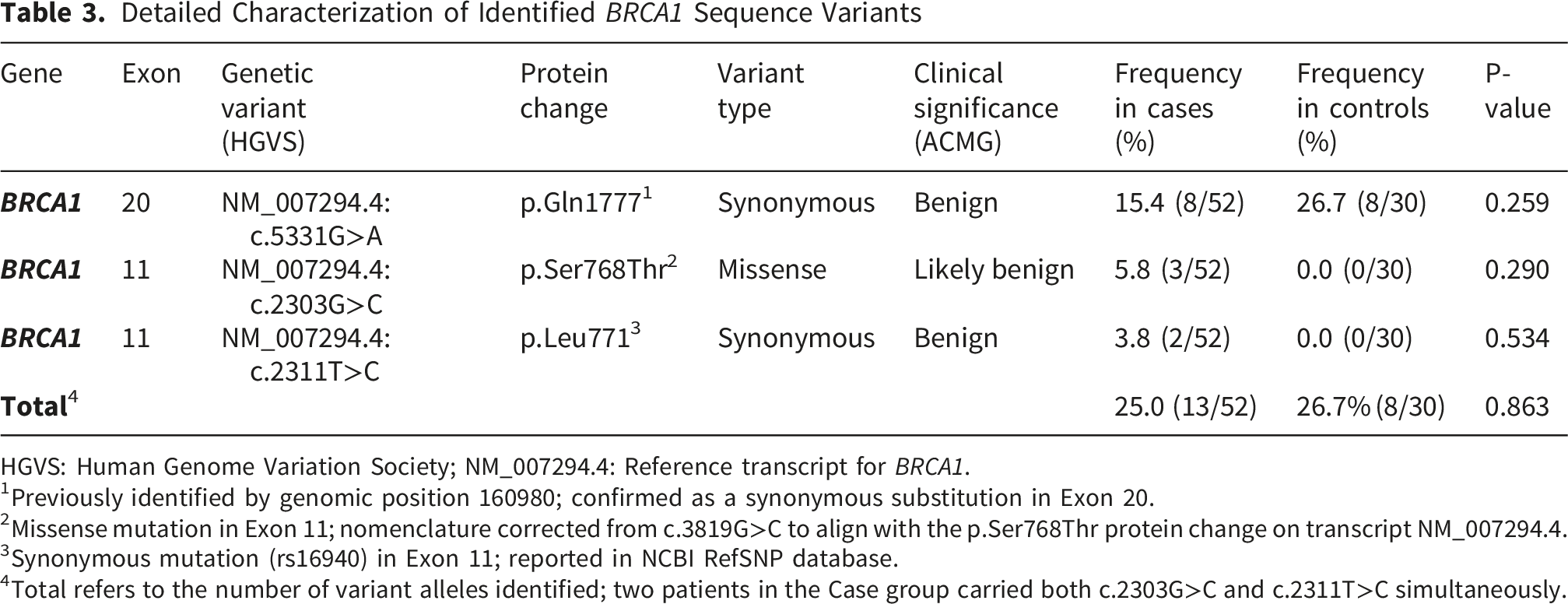
HGVS: Human Genome Variation Society; NM_007294.4: Reference transcript for BRCA1.
1Previously identified by genomic position 160980; confirmed as a synonymous substitution in Exon 20.
2Missense mutation in Exon 11; nomenclature corrected from c.3819G>C to align with the p.Ser768Thr protein change on transcript NM_007294.4.
3Synonymous mutation (rs16940) in Exon 11; reported in NCBI RefSNP database.
4Total refers to the number of variant alleles identified; two patients in the Case group carried both c.2303G>C and c.2311T>C simultaneously.
3.3. The Relationship Between Frequencies of BRCA1 Sequence Variants and Patients’ Clinical and Sociodemographic Characteristics
3.3.1. Association Between BRCA1 Sequence Variants and Sociodemographic Characteristics
Association of BRCA1 Variants With Sociodemographic and Clinicopathologic Characteristics
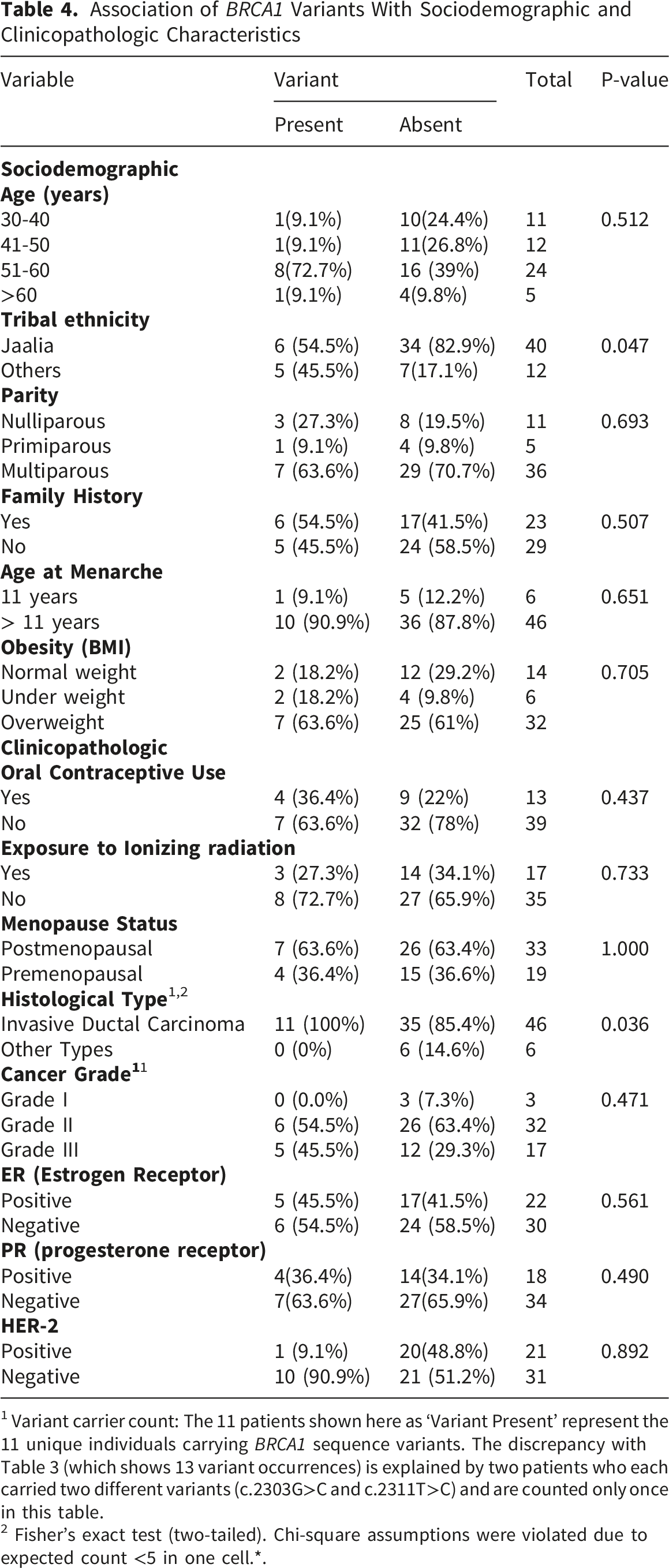
1 Variant carrier count: The 11 patients shown here as ‘Variant Present’ represent the 11 unique individuals carrying BRCA1 sequence variants. The discrepancy with Table 3 (which shows 13 variant occurrences) is explained by two patients who each carried two different variants (c.2303G>C and c.2311T>C) and are counted only once in this table.
2 Fisher’s exact test (two-tailed). Chi-square assumptions were violated due to expected count <5 in one cell.*.
3.3.2. Association Between BRCA1 Sequence Variants and Clinical Characteristics
The study found a strong association between IDC and breast cancer sequence variants (100%, 11/11 variant carriers with IDC diagnosis), with no significant association between histological type and variant frequency (p= 0.0. Variants were more common in grade II cancers (54.5%), estrogen receptor (ER)-negative cases (54.5%), and progesterone receptor (PR)-negative cases (63.6%).
3.4. Detailed Characterization of BRCA1 and BRCA2 Sequence Variants
PCR amplification confirmed the expected product sizes for BRCA1 exon 11 fragments (239 bp and 224 bp), BRCA1 exon 20 (401 bp and 451 bp), and BRCA2 exon 11 using a 100 bp molecular ladder (Figure 1). Sequencing followed by multiple sequence alignment identified three BRCA1 sequence variants across cases and controls (Figure S2 and Figure S3). Sequence analysis was performed using BioEdit software, with alignment against the reference transcript NM_007294.4. Variants were described according to HGVS (Human Genome Variation Society) nomenclature standards. PCR amplification of BRCA1 and BRCA2 exons in breast cancer patients. (A)- BRCA1 exon 11-D (224 bp); lane M: 100 bp DNA ladder; Lanes 1–4: samples from patients. (B)- BRCA2 exon 11-B (451 bp); lane M: 100 bp DNA ladder; lanes 1–5: samples from patients. (C)- BRCA1 exon 20-A (401 bp); lane M: 100 bp DNA ladder; lanes 1–2: samples from patients. (D)- BRCA1 exon 11-C (239 bp); lane M: 100 bp DNA ladder; lanes 1–4: samples from patients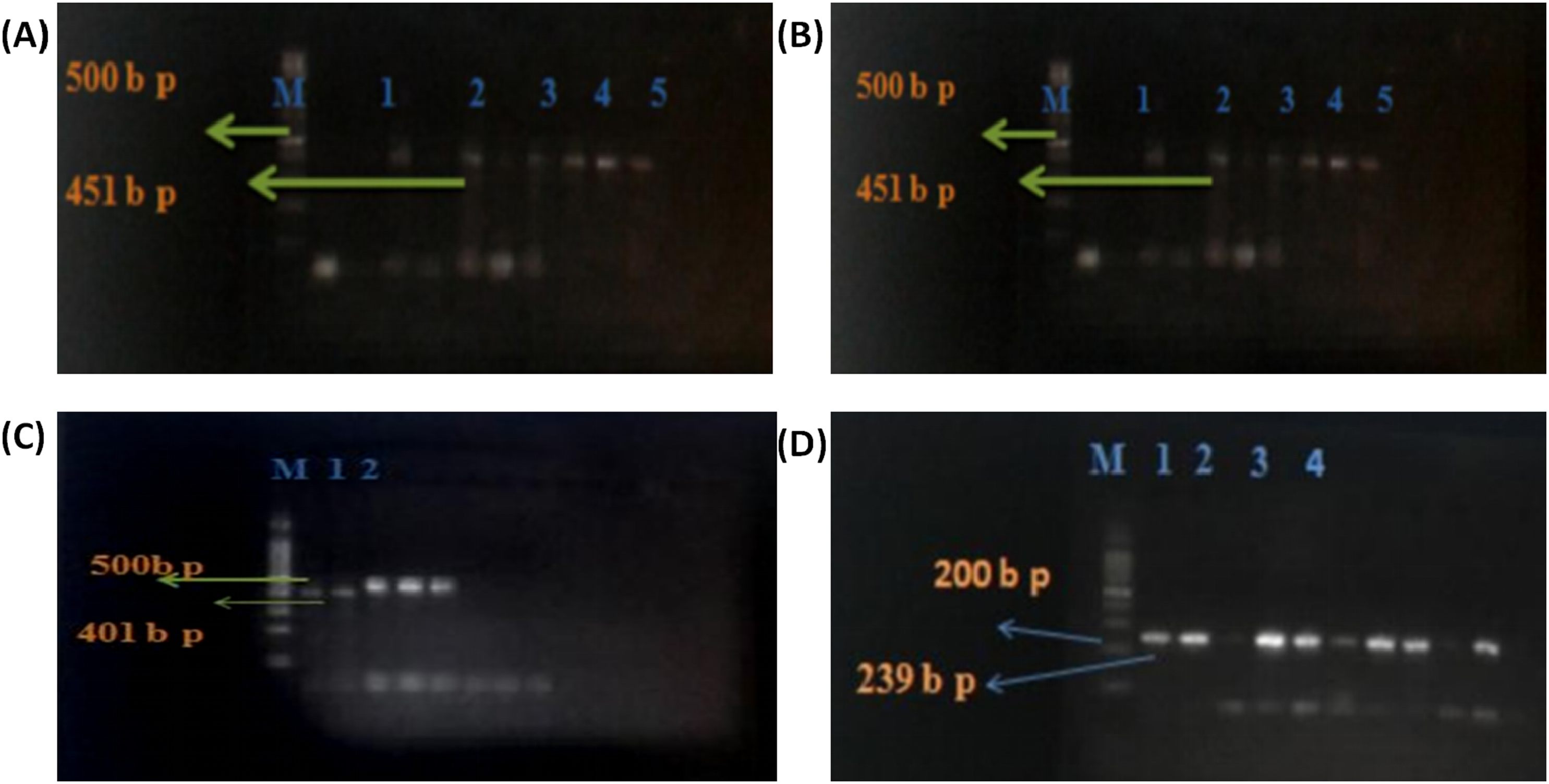
The synonymous variant c.5331G>A (p.Gln1777=) represents a nucleotide substitution that does not alter the encoded amino acid (Figures S2 and S3). The missense variant c.2303G>C (p.Ser768Thr) results in the substitution of serine (AGC) with threonine (ACC) at codon 768 (Figure S4, Figure S5). Amino acid sequences were translated and aligned against the BRCA1 protein reference sequence. Functional impact predictions were assessed using SIFT (Sorting Intolerant From Tolerant), PolyPhen-2 (Polymorphism Phenotyping v2), I-Mutant-3, and PhD-SNP (Table S7). Amino acid physicochemical properties were evaluated using Project HOPE (Figure S6).
The c.2303G>C (p.Ser768Thr) variant was detected in three patients (5.8%) and in none of the controls. The ages of these patients were 45, 55, and 56 years; two reported a positive family history. All three patients belonged to the Jaalia tribe and were diagnosed with invasive ductal carcinoma. The c.2311T>C (p.Leu771=) synonymous variant does not alter the encoded amino acid and is consistent with previously reported SNPs (Single Nucleotide Polymorphisms). No novel pathogenic variants were identified in the analyzed regions. No pathogenic variants were detected in BRCA2 exon 11.
4. Discussion
Breast cancers remain the most frequently diagnosed malignancy among women worldwide, with approximately 2.26 million new cases annually according to IARC Globocan 2022. 1 Despite advances in molecular diagnostics and targeted therapies, they continue to represent a major cause of cancer-related mortality, particularly in low- and middle-income countries where late-stage diagnosis is common.8,9,17,18 This study evaluated selected exons of BRCA1 and BRCA2 in a Sudanese breast cancer cohort alongside demographic and lifestyle factors.
A predominance of cases was observed in urban areas, with 65.4% residing in Shendi. Educational levels varied, and 42.3% of patients were illiterate, which may influence screening awareness and health-seeking behavior. The Jaalia tribe accounted for 76.9% of cases in this cohort. Married women represented the majority of patients, potentially reflecting reproductive and hormonal exposures such as parity and breastfeeding patterns.19,20
Physical inactivity was reported in 88.5% of cases, consistent with evidence linking a sedentary lifestyle to metabolic dysregulation and chronic inflammation. Overweight was present in 61.5% of patients, 21 reinforcing its established role as a modifiable breast cancer risk factor through mechanisms involving estrogen production, insulin resistance, and inflammatory signaling. 22 Smoking prevalence was low (9.6%), consistent with regional epidemiological patterns. 23
At the molecular level, three BRCA1 sequence variants were detected: NM007294.4:c.5331G>A (p.Gln1777=), NM007294.4:c.2303G>C (p.Ser768Thr), and NM_007294.4:c.2311T>C (p.Leu771=). 24 All variants were classified according to ACMG (American College of Medical Genetics) criteria as benign or likely benign. 25 The synonymous variants c.5331G>A and c.2311T>C do not alter the amino acid sequence and are consistent with benign polymorphisms. 26 The missense variant c.2303G>C (p.Ser768Thr) was predicted by in silico tools to have no definitive pathogenic impact and was therefore classified as likely benign. 24 No statistically significant difference in variant frequency was observed between cases and controls. 27
Importantly, no pathogenic variants were identified in BRCA1 exons 11 and 20 or BRCA2 exon 11 in this cohort. 28 Therefore, the detected sequence variants are unlikely to represent major drivers of breast cancer susceptibility in this cohort. However, absence of pathogenic variants in these selected exons does not exclude the possibility of variants in other coding regions, deep intronic regions, or large genomic rearrangements not detectable by Sanger sequencing. 29
We acknowledge the significant limitation of targeted exon screening. While no pathogenic variants were identified in BRCA1 exon 11, exon 20, or BRCA2 exon 11, these findings do not rule out the presence of pathogenic variants in the remaining 41 coding exons of BRCA1 and 26 coding exons of BRCA2. However, given that BRCA1 exon 11 constitutes approximately 60% of the coding sequence and is the most frequent site of pathogenic variants in most studied populations—including North and West African cohorts where founder mutations have been documented—the absence of pathogenic variants in this large exon across patient chromosomes is notable.1,2,5 This observation suggests that the genetic etiology of breast cancer in this specific Sudanese cohort may rely on different BRCA1/2 domains or entirely distinct moderate-penetrance genes (e.g., PALB2, ATM, CHEK2) not assessed in this study.
Global variant frequencies for BRCA1 range from 1.9–13% and for BRCA2 from 2.2–2.7%, depending on population structure and methodology.30,31 No significant association was observed between detected sequence variants and family history, parity, age at menarche, or radiation exposure.32-38 These results are consistent with the benign or likely benign classification of all identified variants and do not support a role for these specific polymorphisms in modulating breast cancer risk in this cohort.
The absence of Variants of Unknown Significance (VUS) in the analyzed exons suggests that the genomic regions examined do not harbor ambiguous variants requiring additional functional studies.39,40 However, if such variants had been identified, their functional significance would require further investigation through cellular and biochemical assays. Larger, more comprehensive genomic analyses are warranted to fully characterize the spectrum of genetic variation in this population.
The relationship between telomere length and BRCA1 variants remains an area of ongoing investigation. 41 Although telomere shortening has been proposed as a marker of genomic instability in BRCA1-associated breast cancers, the biological mechanisms are not fully elucidated.42,43 Further research in larger and ethnically diverse cohorts is required. Overall, expanded genomic approaches incorporating full gene sequencing and structural variant analysis are necessary to better define hereditary breast cancer risk in Sudanese populations.
5. Conclusion
In this Sudanese cohort, targeted sequencing of BRCA1 exons 11 and 20 and BRCA2 exon 11 did not identify pathogenic variants. The detected BRCA1 sequence variants were classified as benign or likely benign according to ACMG/AMP criteria and showed no significant association with breast cancer status. Notably, the absence of pathogenic variants in BRCA1 exon 11—the largest exon and a well-established mutational hotspot in diverse populations—suggests that the genetic architecture of breast cancer susceptibility in Sudanese patients may differ from that observed in other African and global cohorts. Comprehensive genomic studies incorporating full gene sequencing, copy number variant analysis, and inclusion of diverse Sudanese ethnic groups are required to fully elucidate the spectrum of hereditary breast cancer risk in this population.
6. Limitations
This study has several limitations that warrant careful interpretation. First, due to resource constraints, sequencing was restricted to three specific exonic regions. Consequently, the reported absence of pathogenic variants applies strictly to the analyzed amplicons and cannot be extrapolated to the entire BRCA1 and BRCA2 genes. Variants located in other coding regions, deep intronic regions, promoter regions, or large genomic rearrangements were not assessed and may contribute to breast cancer susceptibility in this population. Second, the sample while appropriate for a pilot case-control comparison of specific regions, is underpowered to detect rare variants in a genetically diverse nation like Sudan. Third, the study cohort was drawn predominantly from a single geographic region (Shendi, Northern Sudan) and a single ethnic group (Jaalia tribe, 76.9%), which limits generalizability to other Sudanese populations. Despite these constraints, this report provides the first publicly available sequencing data for these specific exonic regions in an indigenous Sudanese population, establishing baseline allele frequencies for observed benign polymorphisms and demonstrating that BRCA1 exon 11—the largest exon and a global mutational hotspot—does not appear to harbor recurrent pathogenic variants in this specific sub-population.
Supplemental Material
Supplemental material - Low Frequency of Pathogenic Variants in BRCA1 Exons 11/20 and BRCA2 Exon 11 Suggests Divergent Mutational Hotspots in Sudanese Breast Cancer Patients: A Case-Control Study
Supplemental material for Low Frequency of Pathogenic Variants in BRCA1 Exons 11/20 and BRCA2 Exon 11 Suggests Divergent Mutational Hotspots in Sudanese Breast Cancer Patients: A Case-Control Study by Hadia Abass Eltaib Ahmed, Babbiker Mohammed Taher Gorish, Ghanem Mohammed Mahjaf, Waha Ismail Yahia Abdelmula, Alsmawal A. Elimam, Hisham N. Altayb, Mona Dawood, Asma Al-Ameer M. Zeen, Tibyan Abd Almajed Altaher, Emad Bakri Abass Ahmed, Rashid Eltayeb Abdalla in Breast Cancer: Basic and Clinical Research
Supplemental Material
Supplemental material - Low Frequency of Pathogenic Variants in BRCA1 Exons 11/20 and BRCA2 Exon 11 Suggests Divergent Mutational Hotspots in Sudanese Breast Cancer Patients: A Case-Control Study
Supplemental material for Low Frequency of Pathogenic Variants in BRCA1 Exons 11/20 and BRCA2 Exon 11 Suggests Divergent Mutational Hotspots in Sudanese Breast Cancer Patients: A Case-Control Study by Hadia Abass Eltaib Ahmed, Babbiker Mohammed Taher Gorish, Ghanem Mohammed Mahjaf, Waha Ismail Yahia Abdelmula, Alsmawal A. Elimam, Hisham N. Altayb, Mona Dawood, Asma Al-Ameer M. Zeen, Tibyan Abd Almajed Altaher, Emad Bakri Abass Ahmed, Rashid Eltayeb Abdalla in Breast Cancer: Basic and Clinical Research
Footnotes
Acknowledgments
The authors acknowledge Al-Neelain University in Khartoum’s generous help in conducting this study. Also, we would like to thank the Radiation and Isotope Center in Shendi City for their approval of this work and cooperation in sample collection. We appreciate the contribution of Shendi University, Sudan. Thanks to all of the breast cancer patients who agreed to participate in my study; without them, this study would not have been possible to see the light.
Ethical Considerations
The study was approved by the Ethical Committee of El-Mek Nimr Hospital, approval No. 2/9/2013. All participants were informed about the study’s objectives, and written informed consent was obtained from each participant. All procedures involving human subjects were conducted in accordance with institutional and national ethical standards and with the 1964 Helsinki Declaration and its later amendments.
Consent to Participate
Informed consent was obtained from all subjects involved in the study.
Authors’ contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Supplemental Material
Supplemental material for this article is available online.
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.