
Abstract
Backgrounds:
About 25% to 30% of estrogen receptor (ER)-positive breast cancer patients develop resistance to endocrine therapy. Human epidermal growth factor receptor 2 (HER2) has been shown to cooperate with several growth factors that regulate cellular energy metabolism, including the insulin-like growth factor 1 receptor (IGF-1R).
Objective:
As the first-line therapy for type 2 diabetes mellitus (T2DM) patients, metformin is widely known to inhibit the metabolic reprogramming of cancer cells. This study aims to investigate metformin’s efficacy in inhibiting endocrine resistance related to genes regulating energy metabolism in both ER-positive and ER-negative breast cancer cell lines under hyperglycemic conditions.
Design and methods:
MDA-MB-361 (ER-positive, HER2-positive) and SKBR3 (ER-negative, HER2-positive) cancer cell lines were used to represent ER status. Cell viability and cell survival rate were measured using the colorimetric assay of Cell Counting Kit-8. All mRNA levels were quantified using real-time quantitative polymerase chain reaction preceded by reverse transcription. A P value of <.05 was considered statistically significant.
Results:
Unlike MDA-MB-361, SKBR3 were found to acquire resistance upon metformin treatment in hyperglycemic conditions. Moreover, the mRNA expression of IGF-1R and its downstream signaling, such as the mammalian target of rapamycin (mTOR), was not affected by metformin. Meanwhile, the mRNA expression level of ribosomal S6 kinase 1 (S6K1) was upregulated, whereas forkhead box O1 (FOXO1) was downregulated after metformin treatment in hyperglycemic conditions.
Conclusions:
This preliminary study suggests that an alternative pathway of metformin resistance may exist in the absence of ERα. Therefore, relying solely on metformin may be inadequate to inhibit the aggressiveness of breast cancer cells.
Plain Language Summary
Around 25% to 30% of breast cancer patients with estrogen receptor (ER)-positive tumors become resistant to hormone therapy. This study explores whether metformin, a drug commonly used for type 2 diabetes, can counteract this resistance by affecting genes linked to energy metabolism. The research focused on both ER-positive (MDA-MB-361) and ER-negative (SKBR3) breast cancer cell lines under high-glucose conditions. Results showed that although metformin inhibited the growth of ER-positive cells, it surprisingly promoted resistance in ER-negative cells. The expression of insulin-like growth factor 1 receptor (IGF-1R) and its downstream signals like mammalian target of rapamycin (mTOR) remained unaffected by metformin. However, metformin did alter the expression of other genes related to energy metabolism, suggesting that a different resistance pathway might exist in ER-negative cases. In conclusion, this early study implies that relying solely on metformin might not be sufficient to combat the aggressiveness of breast cancer cells, particularly in cases lacking ERα. More research is needed to understand alternative pathways and develop more effective strategies against resistance.
Introduction
Two-thirds of breast cancers are hormone-dependent marked by the presence of estrogen receptor α (ER-positive). The treatment for this type of breast cancer is typically antiestrogen agents, such as tamoxifen and aromatase inhibitors. 1 However, resistance to endocrine treatments occurs in approximately half of ER-positive patients, indicating that these cancer cells exploit multiple pathways to evade targeting of the ER. 2 For example, the involvement of growth factor receptor pathways, such as human epidermal growth factor receptor 2 (HER2), exaggerates the endocrine therapy resistance.3,4 Earlier studies have demonstrated that an increase in HER2 expression is accompanied by an elevated glycolytic rate in tamoxifen-resistant cells, providing a rapid energy source for tumor growth. 5
Numerous studies have confirmed that the presence of excessive glucose in type 2 diabetes mellitus (T2DM) could trigger metabolic reprogramming of cancer cells, promoting their survival and correlating with an increased risk of breast cancer recurrence.6-9 The chronically elevated blood glucose levels in T2DM patients promote the upregulation of growth factors signaling, including insulin and insulin-like growth factors (IGFs), to enhance glycolytic capacity.10,11 Indeed, IGF-1 also plays roles in mammary terminal end bud and ductal formation and maintenance of the mammary gland, and it is excessively expressed in aberrant breast cancer cells.12,13 Moreover, phosphorylated insulin-like growth factor 1 receptor (IGF-1R) and insulin receptor (IR) are markedly expressed in human breast cancers of all subtypes regardless of the ER status, contributing to poor prognosis in patients. 14
Studies have shown that the presence of ERα largely upregulates the IGF signaling, and inevitable cross-talks occurs between ERα and IGF-1R in breast cancers. 15 The cross-talks involve the mitogenic downstream signal transduction pathways of the mammalian target of rapamycin (mTOR), where the ribosomal S6 kinase 1 (S6K1) activity as a downstream target of mTOR is required to increase cell proliferation through hormone-independent pathways.4,16,17 Numerous studies have reported that S6K signaling positively correlates with the resistance of ER-positive and ER-negative breast cancer cells to endocrine drugs.18-20 Although signaling through IGF-1R has been complex and controversial, collective studies showed that the inhibition of insulin and IGF-1R stimulates the chemotherapy’s sensitivity and reduces hormone independence in breast cancers.21,22
Considering the intricate interplay between IGF signaling and breast cancer resistance, exploring alternative therapeutic approaches becomes crucial. In this context, targeting glucose metabolism, a strategy with variable responses among patients, is an area requiring further investigation. Metformin (1,1-dimethyl biguanide hydrochloride), the first-line therapy for T2DM patients, has been confirmed to reduce the risk of cancer progression.23-25 However, the mechanism underlying metformi-n’s antiproliferative effect in cancer remains unclear. 26 One potential mechanism could be the activation of adenosine monophosphate–activated protein kinase (AMPK) phosphorylation pathway to inhibit mTOR signaling pathways and subsequently reduce protein synthesis and cell growth. 27 A notable example is through the phosphorylation of Insulin Receptor Substrate-1 (IRS-1) by AMPK, where Akt signaling is reduced, and mTOR pathway is indirectly inhibited. 27 Notably, AMPK is responsible for regulating energy homeostasis and activating many genes involved in cellular metabolism, such as the expression of tumor suppressor genes. 28
The forkhead box O (FOXOs) family is one of the tumor suppressor genes that plays a central role in regulating diverse cellular functions. 29 Without the presence of growth factors, FOXO proteins localize to the nucleus. 30 These proteins play a role in cell apoptosis, cell cycle arrest, autophagy, and stress resistance by upregulating the target genes responsible for those occurrence. 30 However, high expression of growth factors signaling causes the nuclear exclusion of FOXOs to the cytosol and subjects them to the ubiquitin-proteasome degradation system.31,32 Compared with normal mammary epithelial cells, decreased FOXOs expression was observed in breast cancer cells. 33 Interestingly, a recent suggestion shows that co-expression of ER is needed for FOXOs to function as a tumor repressor. 34 Therefore, this study was conducted to determine the molecular mechanism underlying metformin’s antiproliferative action by involving the genes that regulate energy metabolism and its correlation with ERα status under hyperglycemic conditions.
Materials and Methods
Cell cultures
The human breast cancer cell line MDA-MB-361 (ER-positive, HER2-positive) and SKBR3 (ER-negative, HER2-positive) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA; MDA-MB-361 ATCC HTB-27; SKBR3 ATCC HTB-30). The cells were cultured in glucose-free Dulbecco’s modified Eagle’s medium (DMEM, glucose-free 11966025; Thermo Fisher Scientific, MA, USA) supplemented with either 5 mM or 15 mM glucose. Both medium were supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA), 1% sodium pyruvate (Thermo Fisher Scientific), and 1% penicillin-streptomycin (P/S; Sigma-Aldrich, St. Louis, MO, USA). Culture was conducted at 37°C in a humidified incubator containing 95% air and 5% CO2.
Cell proliferation assay
Cell viability and survival was determined in a 96-well plate, with 1000 cells seeded per well containing DMEM with 10% FBS, 1% sodium pyruvate, and 1% P/S, and incubated for 48 hours. Next, the medium was replenished with fresh medium containing various glucose concentrations (5, 10, 15, and 25 mM glucose) or the same medium containing metformin (5, 10, and 15 mM) (MP Biomedicals, LLC, Santa Ana, CA, USA) and cells were incubated for another 48 hours. The cell viability and survival were measured by performing the CCK-8 assay (Dojindo, Japan) as per the manufacturer’s instructions. Absorbance was measured at 450 nm using a Tecan microplate reader. All conditions were studied with both technical and biological triplicates, meaning that each experimental condition was performed in 3 independently cultured samples (biological replicates), and each sample was measured 3 times (technical replicates). The absorbance values were used to calculate the percentage of surviving cells for each group, and the results were normalized to controls. Control cultures were defined as 100% survival.
Quantitative reverse transcriptase-polymerase chain reaction
A total of 1 µg of total RNA was extracted from the cells and used as a template for reverse transcription using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Inc, Foster City, CA, USA). All amplifications were perform-ed using the SYBR Green PCR Master Mix (Applied Biosystems, Inc). There were 3 replicates in each gene combination (FOXO1, IGF-1R, mTOR, S6K1, and 18S). The assay was performed according to the manufacturer’s protocol provided with the StepOnePlus Real-Time System (Applied Biosystems, Inc). The reaction was performed under the following conditions: 50°C for 2 minutes, 95°C for 2 minutes, and 40 cycles of 95°C for 15 seconds, 60°C for 15 seconds, and 72°C for 1 minute. Melting curve analysis of every reaction was performed after each cycle. Relative mRNA levels were calculated using the comparative cycle threshold (Ct) method (ΔΔCt). The Ct values for the housekeeping gene (18S) were subtracted from the Ct values of the genes listed in Table 1 to obtain the ΔCt value. Then, 2−ΔΔCt was calculated and divided by the value of a control sample to determine the relative mRNA levels. Reported values are the means and standard errors of the 3 biological replicates.
Primers (Fasmac, Japan) used for qRT-PCR analysis.

Statistical analysis
Statistical analysis was performed using GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA). Data from 2 different groups were presented as mean values ± standard deviation (SD) or standard error of the mean (SEM). One-way analysis of variance (ANOVA) with the Dunnett multiple comparison test was used to determine the significance of differences in cell proliferation rate. Two-way ANOVA with the Bonferroni multiple comparison test was performed to determine the significance of differences in glucose-metformin treatment and to check the possible difference between 2 different cell types. The 2-tailed Student t-test was used to determine the significance of differences in quantitative reverse transcriptase-polymerase chain reaction results between nontreatment and metformin treatment in each glucose concentration. A P value of <.05 was considered statistically significant.
Results
The increase of glucose concentration has contradictory effect on cell proliferation in MDA-MB-361 and SKBR3 breast cancer cells
We investigated cell proliferation rate at various glucose concentrations (0-25 mM) by comparing the viability of breast cancer cells under the presence of 5 mM glucose (G5; euglycemia), chosen to represent a physiological glucose level (Figure 1). G0 (0 mM glucose) was represented as an absence of glucose. G10 (10 mM glucose) and G15 (15 mM glucose) were used to describe hyperglycemic conditions. Although glucose levels of >17 mM/L blood are incompatible with cell viability, we tested G25 (25 mM) as an extreme condition. The cell proliferation rate was inhibited on both cells at 0 mM glucose concentration. However, we found a considerable increase in cell viability only in ER-positive cells (MDA-MB-361) upon the increase in glucose concentrations. Meanwhile, ER-negative cells (SKBR3) showed a dramatic decrease in cell viability at concentration above 10 mM. These findings showed different optimum glucose concentrations for breast cancer cells with different ER statuses. Therefore, we used glucose 5 mM as the normal glucose conditions and 15 mM as the high-glucose conditions for subsequent experiments as we discovered the cell viability in both cells was similar.
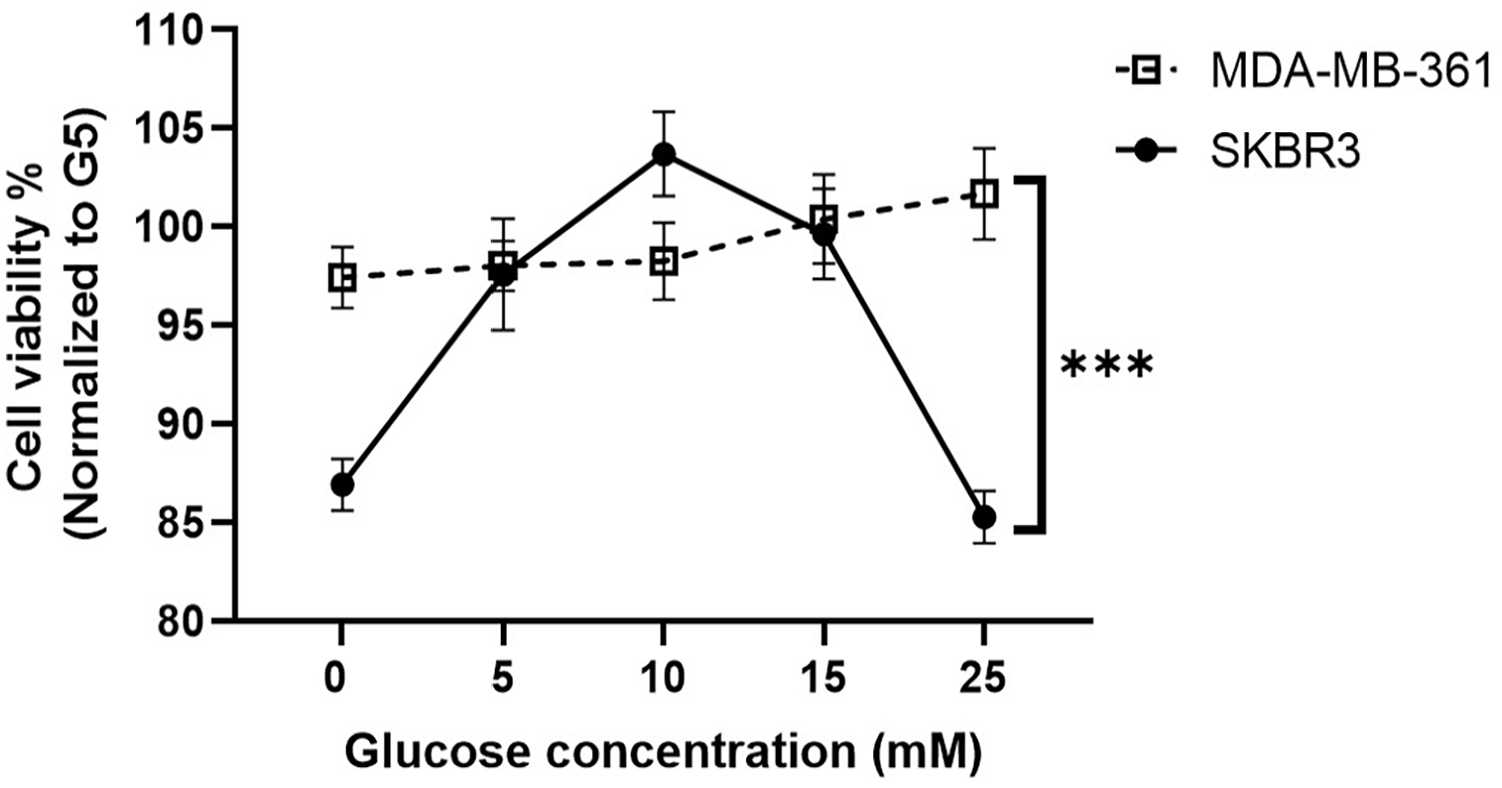
Increased glucose levels promote cell proliferation in MDA-MB-361 cells, but not in SKBR3 cells. Both cells were cultured in different levels of glucose (0, 5, 10, 15, 25 mM) for 2 days. Cell viability was measured using CCK-8 assay. MDA-MB-361 cells thrived in high-glucose medium, whereas SKBR3 cell numbers decreased above 10 mM glucose concentration. Data represented as mean ± SEM from 3 independent experiments. The statistically significant difference of ***P < .001 was analyzed using 2-way ANOVA.
Metformin effectively inhibits cell proliferation in MDA-MB-361 but not in SKBR3 breast cancer cells under hyperglycemic conditions
Several studies have reported that metformin showed a significant inhibitory effect at a concentration of 20 mM on breast and gastric cancer, even though the steady-state therapeutic dose of metformin for humans is in the approximate range of 6 to 30 µM.35-37 We analyzed the antiproliferative effects of metformin at concentrations of 5 and 15 mM to justify the high glucose used in the experiment in MDA-MB-361 and SKBR3 cells at G5, G10, and G15. 38 The results showed greater inhibition in response to metformin in a dose-dependent manner upon the increase in glucose concentrations for MDA-MB-361 cells (Figure 2A). In contrast, metformin inhibition response only occurred on the G5 condition in SKBR3 cells, whereas G10 and G15 conditions showed resistance toward metformin treatment (Figure 2B). Interestingly, metformin resistant was observed in another ER-positive breast cancer cells MCF-7, where it was owed to long-term exposure to metformin when ERα no longer responded.39,40 Thus, it suggests that the regulation of ERα might be required for metformin sensitivity, especially under hyperglycemic conditions.

Glucose availability influences metformin treatment. Cell survival rate was measured using CCK-8 assay after 48 hours incubation with different glucose concentrations (5, 10, 15 mM) followed by another 48 hours of different metformin (0, 5, 15 mM) treatment. (A) Metformin induced cell survival inhibition in a dose-dependent manner across all glucose concentrations in MDA-MB-361 cells. (B) Metformin inhibited cell proliferation only in 5 mM glucose concentration, whereas high-glucose condition desensitized metformin treatment in SKBR3 cells. Data represented as mean ± SD from 3 independent experiments. The statistically significant difference of ***P < .001 and ****P < .0001 was analyzed using 2-way ANOVA between glucose 5 mM (G5) and glucose 15 mM (G15).
Metformin downregulates energy metabolism of insulin-like growth factor 1 receptor/mammalian target of rapamycin/S6 kinase 1 signaling in MDA-MB-361 breast cancer cells under hyperglycemic conditions
To unravel the underlying mechanism of the antiproliferative effect of metformin, we examined gene expression changes of IGF-1R, mTOR, and S6K1 under hyperglycemic conditions upon the addition of metformin. Evidence shows that in the states of insulin resistance, IGF-1 mediates the regulation of glucose metabolism. 41 However, our results showed that metformin consistently suppressed IGF-1R expression as glucose concentration increased in MDA-MB-361 cells (Figure 3A). Interestingly, it was also followed by a considerable downregulation of the master metabolic regulator of mTOR/S6K1 expression in MDA-MB-361 cells that only appeared in G15. Meanwhile, this inhibition effect of metformin failed to induce at high-glucose conditions in SKBR3 cells as we discovered a significant reduction only at G5 (Figure 3B), which indicates a resistance might occur in the excessive glucose level when the ER does not exist, or there is a lack of response. A recent study has confirmed that resistance in hormone-dependent breast cancers was followed by the reduction of ERα phosphorylated on Ser167. 42 Although a previous study has shown that the crosstalk between ERα and IGF-1R correlates with antiestrogen resistance in MDA-MB-361 cells, the interplay between IGF-1R and another growth factor, such as HER2, is ineluctable to induce cell survival in the absence of ER.43-45 Moreover, a high-glucose level protects cells against metformin cytotoxicity by increasing the glycolytic process. 46 Therefore, we found that metformin has a more profound effect on altering cancer cell metabolism in MDA-MB-361 cells due to its ability to simultaneously influence multiple pathways related to cell growth and energy metabolism.

mRNA fold change with respect to IGF-1R, mTOR, and S6K1 in MDA-MB-361 and SKBR3 cells treated with metformin under the presence of 5 and 15 mM glucose. After treatment, cells were lysed and mRNA extracts were analyzed using qRT-PCR. qRT-PCR results showing mRNA fold change with respect to IGF-1R, mTOR, and S6K1 in (A) MDA-MB-361 and (B) SKBR3 cells treated with 5 mM metformin in different glucose conditions (glucose 5 mM [G5]; glucose 15 mM [G15]). Data represented as mean ± SEM from 3 biological experiments with 3 technical replicates per each experiment. The statistically significant difference of *P < .05; **P < .005; ***P < .0005; ****P < .0001 was analyzed using unpaired T-test.
Metformin increases forkhead box O1 mRNA level in MDA-MB-361 but not in SKBR3 breast cancer cells under hyperglycemic conditions
To extend the underlying mechanism from the above results, we assessed the FOXO1 response to metformin-induced apoptosis. The results showed an upward trend of FOXO1 mRNA levels both at G5 and G15 in MDA-MB-361 cells after metformin treatment (Figure 4A). On the contrary, the trend conversely occurred in SKBR3 cells as we found a significant decrease of FOXO1 mRNA fold change both at G5 and G15 after metformin treatment (Figure 4B). Metformin has been shown to enhance the activity of FOXO1 by activating AMPK, suggesting that metformin is an FOXO1 activator that induces cell apoptosis.47,48 Interestingly, we only found this significant effect in MDA-MB-361 cells, whereas this expression was downregulated in SKBR3 cells. Forkhead box O1 is majorly regulated through Akt-mediated phosphorylation, a downstream signal of IGF-1R. 49 This finding supported the previous data showing that, in the absence of ER, IGF-1R may interact with other factors to stimulate cell survival and induce resistance to the treatment in hyperglycemic conditions.

mRNA fold change of FOXO1 in MDA-MB-361 and SKBR3 cells treated with metformin under the presence of 5 and 15 mM glucose. After treatment, cells were lysed and mRNA extracts were analyzed using qRT-PCR. qRT-PCR results showing FOXO1 mRNA fold change in (A) MDA-MB-361 and (B) SKBR3 cells treated with 5 mM metformin in different glucose conditions (glucose 5 mM [G5]; glucose 15 mM [G15]). Data represented as mean ± SEM from 3 biological experiments with 3 technical replicates per each experiment. The statistically significant difference of *P < .05; **P < .005; ***P < .0005; ****P < .0001 was analyzed using unpaired T-test.
Discussion
Several epidemiological studies have reported metformin as a potent anticancer drug due to its ability to manipulate cancer cell metabolism.50-54 In breast cancers alone, the role of metformin has been well studied.39,55 However, metformin resistance occurred in breast cancer cells expressing ERα by involving regulatory genes in energy metabolism.56-58 In this study, we aimed to examine the effect of metformin in 2 different ER statuses (ERα-positive and ERα-negative) under high-glucose conditions at the mRNA expression level of the energy metabolism genes, aiming to understand the impact of metformin on energy metabolism regulation in the context of ERα-positive breast cancer cells.
Interestingly, we found that the proliferation trends of ER-negative SKBR3 cells were different in high-glucose levels compared with ER-positive MDA-MB-361 cells, which showed a growth suppression effect upon the increase of metformin concentrations. It has been suggested that hyperglycemia could maneuver the response of metformin-induced AMPK signaling, whereby cancer cells use insulin/IGF-I signaling to favor the growth-promoting signals through increased glycolysis rate instead of oxidative phosphorylation.46,59,60 Similar to the previous study, we confirmed that metformin effectively inhibits IGF-1R mRNA expression levels regardless of glucose conditions, except in ER-negative SKBR3 cells under high-glucose conditions, suggesting the interplay between ER status and IGF-1R might propose a different metabolic regulation depending on glucose concentration. 61 As a Thr172 AMPK activator, metformin mainly blocks mTOR signaling via IGF-1R and its downstream signals related to the PI3K/Akt/mTOR pathways by activating tuberous sclerosis 2 (TSC2). 59 A recent study found that mTOR suppression by metformin is also induced via a Rag GTPase-dependent manner, which explains another additional AMPK-independent mechanism. 62 Nevertheless, our result showed a different inhibition effect of IGF-1R and mTOR by metformin depending on ER statuses, in which ER-negative SKBR3 cells showed resistance toward metformin under hyperglycemic conditions.
This study also observed that numerous cancer-promoting pathways in ER-positive MDA-MB-361 cells are targeted by metformin. A previous study showed that pS6K1 overexpression in ER-positive breast cancer is positively correlated with the development of endocrine drug resistance. 26 In line with the previous IGF-1R/mTOR expression levels, the mTOR/S6K1 signaling was also significantly inhibited upon metformin treatment both in normal and high-glucose con-ditions in ER-positive MDA-MB 361 cells but not in ER-negative SKBR3 cells under high-glucose condition. It indicates that this signaling is activated mainly by ER, facilitating the crosstalk between ER and IGF-1R.21,63 Interestingly, information can be inferred from Figure 2 that the interplay between glucose abundance and S6K1 expression in ER-positive MDA-MB-361 cells happened the other way around, where S6K1 activated ERα and promoted cell proliferation when not inhibited by metformin, achieved through direct phosphorylation of ERα on Ser167 to induce ER-dependent cell proliferation. 18 With metformin presence, metformin inhibited S6K1 through dephosphorylation on Ser473, subsequently blocking ERα activation. 64 Although collective studies have reported that metformin use was augmented in ER-positive breast cancer cells, its anticancer effect also prominently showed in HER2-positive breast cancer cells, as it acts through the same downstream signaling pathway.65-67 Therefore, metformin more significantly works in hormone receptor-positive and growth factor signaling as the interaction of signaling cascade of PI3K/AKT/mTOR requires these 2 to promote cell growth and survival.
Furthermore, we also discovered an increase in FOXO1-mediated apoptosis expression, especially in ER-positive MDA-MB-361 cells, indicating an additional mechanism underlying metformin’s inhibition of cancer cell growth. The activation of IGF-1R signaling has been reported to mediate FOXO1 inactivation through Akt-dependent phosphorylation, marked by the translocation of FOXO1 from the nucleus to the cytoplasm, a condition common in most cancers.49,68 Nevertheless, nuclear localization of FOXO1 produces an antiproliferative effect by inducing pro-apoptotic genes and insulin-like growth factor-binding protein 1 (IGFBP1), yet the presence of ERα normally suppresses its function in a ligand-dependent manner.48,69 As the IGF-1R has been downregulated by metformin in ER-positive MDA-MB-361 cells, we presume that metformin might indirectly act toward ERα signaling, allowing the FOXO1-mediated transcription of cell cycle arrest and apoptosis. 70 Although previous findings have demonstrated metformin inhibition on estrogen receptor machinery, long-term use of metformin can lead to irreversible cell resistance to endocrine treatment, such as tamoxifen. 39 Recent studies have shown that tamoxifen-resistant breast cancer cells, characterized by insufficient IGF-1R, require this receptor for phosphorylation-dependent ERα activity to induce FOXO1 expression. 43 This suggests that the duration of metformin treatment should be carefully monitored in combination with tamoxifen. Similar to tamoxifen-resistant cells, SKBR3 cells also feature low IGF-1R levels and even ER-negative status, resulting in insufficient induction of FOXO1 expression levels due to the lack of ERα. Surprisingly, we found that a high-glucose condition is required for metformin-induced FOXO1 expression via IGF-1R in ER-negative SKBR3 cells. This phenomenon is similar to the previous study on tamoxifen-resistant cells, in which IGF-1R is necessary to induce FOXO1-mediated IGFBP1 transcription. 43
We observed that ER-negative SKBR3 cells developed resistance to metformin with an increase in glucose concentration. A plausible mechanism for cytotoxicity resistance appears to be linked to nutrient availability. This correlation aligns with a prior study that demonstrated upregulation of pyruvate kinase M2 (PKM2) in SKBR3 cells when glucose was not limiting. 71 The increased expression of PKM2 is influenced by IGF-1 signaling, facilitating the synthesis of metabolites that promote cell growth and proliferation, steering glucose metabolism toward lactate production.72-74
Moreover, another study in SKBR3 cells has identified the involvement of estrogen-related receptors (ERRs), receptor-like ERs that bind to various estrogen elements, inducing tamoxifen resistance. 42 These receptors mimic ER-mediated gene expression in the absence of estrogens and other known ligands, primarily through G Protein-Coupled Receptor GPR30/GPER-1-mediated 17β-estradiol (E2) activation.43,45 Of note, estrogen-related receptor-alpha (ERRα), a master regulator of cellular energy metabolism, has been shown to generate lactate production as an energy source, enabling cells to survive in stress-induced conditions. This adaptation indicates the utilization of anaerobic glycolysis by cancer cells. 75 The shift to lactate production allows cells like SKBR3 to reprogram their metabolism. In addition, the interaction between HER2 and IGF-1R in ER-negative cells stimulates ERRα activity by activating peroxisome proliferator–activated receptor-gamma coactivator-1 beta (PGC-1β). 76 Human epidermal growth factor receptor 2/IGF-1R signaling positively correlates with S6K1 and S6K2/4E binding protein 1 (4EBP1) pathways, inducing hormone-independent growth.20,26
This study possesses several strengths that have yielded new insights into the specific mode of action of metformin as an anticancer agent in high-glucose condition. The incorporation of the interplay between growth factor receptors and hormone receptors in glucose-abundant environment allowed for robust conclusion to be drawn. The quantification of FOXO, serving as a representative of tumor suppressor genes, further elucidated the effect of metformin in cells with different ER statuses. A polarizing effect was observed, providing valuable insights into the impact of metformin on tumor suppressor genes.
Although this study provides a clear perspectives on the impact of metformin in a single ER statuses breast cancer cell lines on the transcriptomic levels of various genes involved in cellular energy metabolism, the quantification of other crucial genes, such as ERα, in multiple ER statuses breast cancer cell lines may be necessary to present a comprehensive view of the interplay. In addition, assessing protein levels of these genes is essential to validate the findings, as the rate of transcription does not always reflect the rate of translation, and post-translational modification may play a key role in regulating the final productions of these genes. Nevertheless, given the crucial role of mRNA as a determinant of disease characteristics and the increasing interest in mRNA-based therapeutics, this study serves as a preliminary exploration to advance our understanding of mRNA’s role in cancer dynamics.
Conclusions
In conclusion, solely hinging on metformin is insufficient to inhibit the aggressiveness of ER-negative SKBR3 breast cancer cells. This suggests a complex interaction between growth factor receptors and hormone receptors contributing to endocrine resistance. Further studies in different ER cell lines are needed to validate the activity of the metabolic genes at the phosphorylation level and explore potential markers regulating energy metabolism in cancer, which could play a role in endocrine resistance.
Research Data
sj-pzfx-3-bcb-10.1177_11782234241240173 – for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions
sj-pzfx-3-bcb-10.1177_11782234241240173 for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions by Andisyah Putri Sekar, Septia Nurmala, Eiji Matsuura, Xian Wen Tan, Ratika Rahmasari and Rani Sauriasari in Breast Cancer: Basic and Clinical Research
Research Data
sj-pzfx-4-bcb-10.1177_11782234241240173 – for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions
sj-pzfx-4-bcb-10.1177_11782234241240173 for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions by Andisyah Putri Sekar, Septia Nurmala, Eiji Matsuura, Xian Wen Tan, Ratika Rahmasari and Rani Sauriasari in Breast Cancer: Basic and Clinical Research
Research Data
sj-pzfx-5-bcb-10.1177_11782234241240173 – for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions
sj-pzfx-5-bcb-10.1177_11782234241240173 for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions by Andisyah Putri Sekar, Septia Nurmala, Eiji Matsuura, Xian Wen Tan, Ratika Rahmasari and Rani Sauriasari in Breast Cancer: Basic and Clinical Research
Research Data
sj-pzfx-6-bcb-10.1177_11782234241240173 – for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions
sj-pzfx-6-bcb-10.1177_11782234241240173 for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions by Andisyah Putri Sekar, Septia Nurmala, Eiji Matsuura, Xian Wen Tan, Ratika Rahmasari and Rani Sauriasari in Breast Cancer: Basic and Clinical Research
Research Data
sj-xlsx-1-bcb-10.1177_11782234241240173 – for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions
sj-xlsx-1-bcb-10.1177_11782234241240173 for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions by Andisyah Putri Sekar, Septia Nurmala, Eiji Matsuura, Xian Wen Tan, Ratika Rahmasari and Rani Sauriasari in Breast Cancer: Basic and Clinical Research
Research Data
sj-xlsx-2-bcb-10.1177_11782234241240173 – for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions
sj-xlsx-2-bcb-10.1177_11782234241240173 for Estrogen Receptor Is Required for Metformin-Induced Apoptosis in Breast Cancer Cells Under Hyperglycemic Conditions by Andisyah Putri Sekar, Septia Nurmala, Eiji Matsuura, Xian Wen Tan, Ratika Rahmasari and Rani Sauriasari in Breast Cancer: Basic and Clinical Research
Footnotes
Acknowledgements
This study was financially supported by Directorate of Higher Education, Ministry of Education, Culture, Research, and Technology, Republic of Indonesia under PDUPT Grant No. NKB-787/UN2.RST /HKP.05.00/2022. We also acknowledge support from Okayama Medical Innovation Center & Department of Cell Chemistry, Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences, Okayama, Japan for the place to conduct the experiment. The authors also thank Kazuko Kobayashi for the technical assistance during the study.
Declarations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.