
Abstract
Biomedical science has made substantial progress toward diagnosing, understanding the pathogenesis, and treating various causative agents of infectious disease. However, novel microbial pathogens continue to emerge, and existing pathogens continue to evolve alternative strategies to thrive in ever-changing environments. Various infectious disease etiological agents originate from animal reservoirs, and several have, over time, acquired the ability to cross the species barrier, altering their host range. Computational approaches in biomedical science capable of analyzing large datasets are invaluable for predicting and monitoring disease outbreaks and their effectiveness is greatly enhanced when integrated with machine learning techniques. The goal of this study is to develop a machine learning model for the prediction of potentially zoonotic organisms, using viral surface proteins that facilitate host cell entry as input data. Sequence data and metadata were obtained from UniProtKB, transformed into a machine-readable format, using frequency chaos game representation and a convolutional neural network model was developed to identify sequence patterns consistent with viruses which infect humans. The model achieves generalized performance of 96.78% accuracy, 0.97 F1 score, and 0.93 MCC (Matthews Correlation Coefficient) on unseen data. The model potentially provides a robust framework for application in early identification of emerging viral threats, supporting public health surveillance and risk mitigation.
Keywords
Introduction
Recently, there has been a marked increase in the emergence and re-emergence of infectious diseases, posing a significant threat to public health.1,2 It is believed that pathogenic etiological agents responsible for the majority of infectious disease outbreaks are zoonotic in origin 3 and zoonotic viruses in particular, are of substantial concern, due to their abundance, diversity, and being notoriously difficult to manage and treat. 4 The virus discovery curve indicates that there is a significant number of viruses which are yet to be discovered.5,6 There is limited knowledge about which of the newly discovered organisms may be pathogenic, and capable of triggering zoonotic events with pandemic potential.5-7
Humans, animals, and pathogens live in a dynamic, interactive, and interconnected environment whereby the health of one affects the other. 1 Human population growth, coupled with activities that result in the encroachment of habitats and perturbation of ecological niches, ultimately facilitate closer interaction between biological entities resulting in close contact between humans and animals (wild and domestic), which may be reservoirs of infectious pathogens.8-10 This proximity relationship is postulated to be one of the drivers of increased emergence and re-emergence of infectious diseases through zoonosis.2,8,11
Zoonotic diseases have been a major economic burden and public health concern on a global scale2,12 and there is a growing need for the development of robust prediction systems to mitigate or even prevent epidemic and pandemic events from occurring. Taking into consideration that zoonosis is a complex phenomenon, it requires multifaceted approaches and the need for predictive methods, to complement current surveillance methods. 13 In addition to traditional surveillance efforts used throughout infectious disease outbreaks,7,14 various models, which harness statistical and ML tools, have been developed to predict cross-species spill-over and transmission dynamics of novel and re-emerging pathogens.15,16 These models incorporate ecological, demographic, and biogeographic data as features for robust algorithm development, to predict potentially zoonotic pathogens, identify existing reservoirs and new potential hosts, and predict zoonotic hotspots using knowledge from disciplines such as ecology and molecular biology.15,17
A number of studies have reported the applications of ML techniques for the analysis of pathogen and host interaction networks. Wardeh et al 17 developed a ML framework using random forests and gradient boosting, trained on mammalian viral traits and network features to predict potential mammalian hosts of known viruses. Shared pathogen networks were also incorporated through graph-based learning to enhance zoonotic reservoir prediction. The study highlighted the role of host phylogeny in pathogen sharing and quantified overlaps between humans and other mammals. Eng et al 18 applied support vector machines to host tropism signatures from avian and human influenza strains to model zoonotic emergence. Qiang and Kou 19 employed deep learning with protein sequence embeddings to predict avian influenza interspecies transmission. Han et al 15 used boosted regression trees and logistic regression with biogeographic and ecological variables to predict rodent reservoir species of undiscovered zoonoses, achieving accuracies in the 90th percentile and identifying over 150 novel hyper-reservoir species. While ecological models assess species crossover events at a macro scale, they lack molecular resolution and often emphasize few hosts or pathogens, limiting generalizable prediction.15,17,20 These limitations motivate the consideration of deep learning models in this study, particularly convolutional neural networks (CNNs), which can automatically extract hierarchical features from genomic and protein data and capture complex nonlinearities in host-pathogen interactions.21,22 Capitalizing on the predictive capabilities of CNNs thus provides a promising avenue for improving zoonotic risk prediction and forms the basis of the present work.
Furthermore, for the analysis of cross-species events, Virus-receptor PPI models have been developed to predict these events.23,24 However, studies are limited by the availability of experimentally derived and validated PPIs, thereby influencing the amount of available input data for ML models to produce robust, translatable, and reproducible models.23-25 In addition, PPI studies rely on defined pre-existing interactions, which may be unable to predict viral host switching in which a previously unknown host receptor is targeted for entry into host cells.26,27 Despite these limitations, various ML approaches have been developed for the analysis of pathogen-host protein interaction networks to predict cross-species events.24,27
Machine learning has also been applied in several genomic surveillance initiatives, with many recent activities focusing on SARS-CoV-2 and Influenza viruses to not only identify emerging variants with the potential future spread, but also predict host range and species susceptibility.28-30 Gussow et al 31 conducted an in-depth molecular analysis of coronaviruses to assess enhanced pathogenicity. Using comparative genomics and ML techniques, the authors identified signatures present in key genomic regions, such as the nucleocapsid protein and the spike glycoprotein, which appear to be associated with higher case fatality rates and host switching. Similarly, coronavirus spike protein sequences were used by Qiang et al 32 to aid the prediction of species cross-over from non-human hosts of this viral taxa, suggesting that SARS-CoV-2 taxonomic relatives may indeed be of concern and should potentially be monitored. UniBind, an artificial intelligence-based framework, integrated protein structure and binding affinity, resulting not only in the efficient prediction of the effects of the binding affinity of SARS-CoV-2 variants to the host receptor but also in the prediction of host susceptibility to these viral variants. 33 A summary of relevant machine-learning models for viral host and spillover prediction is provided in Table 1.
Summary of related machine-learning approaches for viral host prediction and potential spillover assessment.

The study presented in this work advances zoonosis prediction beyond traditional ML approaches that rely on ecological, biogeographic, or trait-based features by focusing directly on viral surface protein sequences. The main contribution of this work is a presentation of a proof-of-concept framework that encodes these sequences using FCGR and applies a CNN classifier to predict whether a virus has zoonotic potential. By demonstrating that FCGR-encoded protein features can capture molecular signatures of zoonosis, this work positions CNNs as a powerful alternative to conventional ML methods, which often struggle to generalize across host-pathogen systems. This contribution provides a scalable, data-driven foundation for more accurate and broadly applicable tools for zoonotic surveillance.
Methods
Data acquisition
Data collected for this study was derived from the UniProtKB Knowledge Database 50 and accessed through the UniProtKB website (https://www.uniprot.org/uniprot/, accessed September 21, 2021). Relevant data table fields were selected and corresponding protein sequences for the data entries, were obtained in FASTA format and the dataset is referred to as KW-1160 throughout. The data consist of a total of 358333 data entries, with exploratory data analysis revealing that the dataset (a) did not exclusively contain entries from viral pathogens and (b) contained 237573 samples with incomplete associated “Virus hosts” metadata. The reporting of this study conforms to the REFORMS statement, 51 the checklist can be found in the supplementary data (Supplemental Table 1). Data used in this study is included in the supplementary information.
Data preprocessing
The KW-1160 dataset preprocessed and cleaned for efficient model development and a Python script was written to automate the cleaning step, requiring 16 CPUs and 32 GB of RAM. The KW-1160 dataset Taxonomic lineage IDs and virus species names were standardized to corresponding ontologies used in the NCBI database using the ete3 toolkit Python package. 52 The virus organism taxonomic super kingdom and family was obtained from the NCBI database, using ete3 toolkit, and added to the existing dataset in appended columns. Microorganisms other than viruses were removed from the KW-1160 dataset.
Addressing missing values
Missing data is a common data quality issue in statistical analysis and ML. Beyond introducing bias into estimates, missing data can also reduce statistical power, distort parameter estimates, limit generalizability, and, in severe cases, render analyses invalid. To address these issues, a variety of techniques have been developed, ranging from simple approaches such as listwise deletion and mean or median computation to more advanced methods like multiple imputation, maximum likelihood estimation, and ML-based imputers.53,54
For viruses with missing “host” information, metadata imputation from external database records was performed in this study. Additional data used for imputation of missing host information in the KW-1160 dataset was obtained from NCBI Virus, 55 Enhanced Infectious Disease Database (EID2) 56 and Virus-Host database, 57 all of which were accessed on September 21, 2021. The data from the external sources was first standardized to use the NCBI taxonomy names, followed by extraction of corresponding taxonomic IDs. The host data was also standardized to match the nomenclature in the KW-1160 dataset, in the format [host name TaxID:ID]. Following standardization, each of the datasets were merged with the KW-1160 dataset, using a left-inner join, such that only samples with matching taxonomic ID would be imputed.
The imputation using NCBI-Virus dataset resulted in a 15% reduction of missing host values, while the imputation using EID2 dataset showed a negligible reduction. The most notable impact of imputation activities was observed when the Virus-Host DB data was used, with an approximate 50% reduction of missing values. This significant reduction is attributed to wider federation of additional information sources such as GenBank, ViralZone, literature surveys, in addition to RefSeq and manual curation of the database. Following imputation of data, the preprocessed KW-1160 dataset used for downstream analysis consists of 317 561 samples.
In addition, a column named Infects human was added to the dataset and contained binary data indicating whether the taxonomic ID for Homo sapiens (9606) was present in the list of viral host names. The rows matching the parameter, labeled “human true,” were considered as positive data, while those which do not match the parameter were considered as negative data and labeled “human false.” The FASTA file containing the protein sequences were mapped to their corresponding samples and the protein names in the KW-1160 dataset were replaced with the protein names in the FASTA headers as a more simplified nomenclature. The headers were then modified to contain the unique entry, protein name, the name of the virus, as well as the infection status, human-true or human-false.
Handling class imbalance and data splitting
To classify the positive and negative dataset, the “Virus hosts” field was designated to indicate whether a viral pathogen was documented to have a human host. Therefore, the primary objective of the model is to predict pathogens with the potential to cross the species barrier and infect humans, and as such, viruses that are reported to successfully infect humans would be classified as positive (with the assumption that they did not originate in the human host) and others as negative. Hence, the problem is formulated as a binary classification problem.
The preprocessed KW-1160 dataset contains class imbalance between the 2 classes, whereby the positive class had substantially more data points (278 791 samples) when compared with the negative class (38 770 samples). A random undersampling (RUS) approach was employed to address the class imbalance problem in the dataset using the imbalanced-learn Python package. 58 Thereafter, the KW-1160 dataset was split into training and test data at a ratio of 80:20 for training and testing purposes, respectively. Also, 20% of the training data was used as a validation set to assess the model generalization before final testing.
Sequence encoding
An R script was written to convert the FASTA protein sequences into machine readable FCGR images using the kaos package. 59 The FCGR chosen was the frequency matrix, with the corners and labels parameters set to false, and default settings are retained for all other parameters. The resulting plots were saved as portable network graphics (PNG) images of 224x224 pixels, at a resolution of 100. Parallel programming coupled with asynchronous programming using the later and parallel R packages, 60 respectively on a computational cluster compute node with 32 CPUs and 40 GB of RAM, was employed for efficient execution of the FCGR conversion process.
Model development
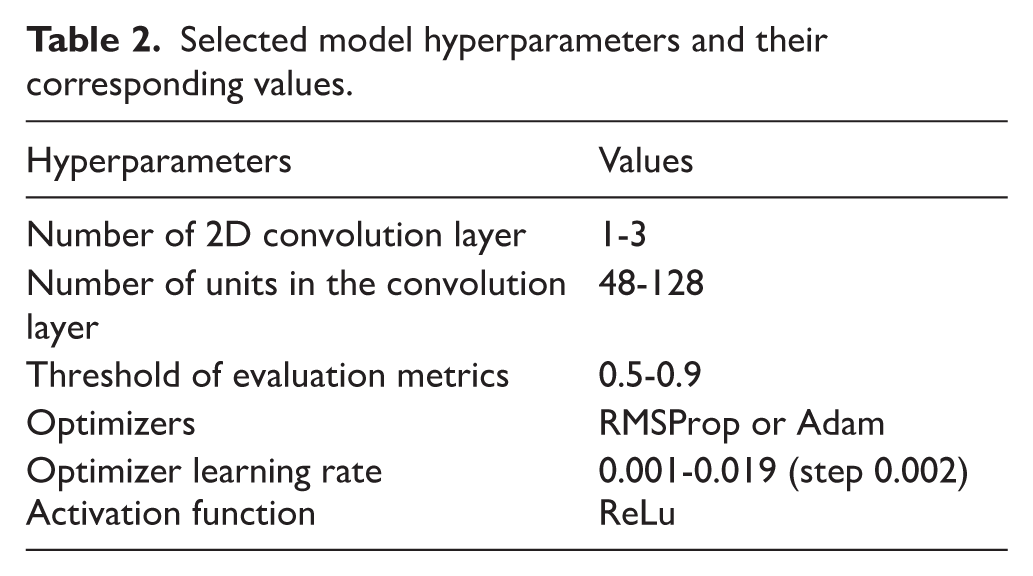
A convolutional neural network (CNN) was used to build the classification model as shown in Figure 1. To obtain optimal parameter values for the number and types of layers in the network, hyperparameter tuning was performed. The Keras-tuner Python package was used to implement Bayesian hyperparameter search. 61 The Bayesian hyperparameter tuning was employed because it efficiently balances exploration and exploitation by learning from previous evaluations, making it more suitable than other tuning strategies such as grid or random search to obtain optimal parameter values suitable to construct the CNN model on the large and computationally intensive KW-1160 dataset. Based on results from the hyperparameter tuning, an optimized CNN model (ie, the model with the best hyperparameter values) with a single convolution and maximum pooling layer was developed using the Keras package and the TensorFlow package in the Python programming language. The CNN model is expected to have captured the key patterns and features in the protein sequence dataset to predict potentially zoonotic organisms. Table 2 presents the selected hyperparameters and their respective values for the developed CNN model. Along with the hyperparameter values provided in Table 2, the search was implemented for 500 trials, 500 combinations of hyperparameters, 3 kernel and pool sizes, and training for 2 epochs per trial, with validation at the end of each epoch.

A depiction of the CNN model architecture created. Each box represents a layer in the model architecture. The arrows represent the flow of information between the layers.
Selected model hyperparameters and their corresponding values.
The InputLayer serves as the entry point of the network, where input data is fed into the model. The Conv2D layer performs 2-dimensional convolution operations to extract image features, while the MaxPooling2D layer reduces dimensionality and retains the most salient features. The Flatten layer then transforms the feature maps from 2 dimensions into a 1-dimensional vector, preparing the data for classification. Finally, the dense layer acts as the output unit, producing the binary classification result.
The rectified linear unit (ReLU) activation function was used in the CNN model development to improve model efficiency by addressing inherent neural network problems such as the vanishing gradient problem.62,63 In addition, sequence similarity was anticipated due to the possibility of conserved regions in transmembrane proteins and high-dimensionality data,64-66 and as such the L2 regularizer was used to prevent the model from overfitting highly correlated data. The sigmoid activation was employed in the final output layer to predict class probabilities for binary classification problems in neural networks.64,66,67
Model training was implemented on an NVIDIA graphics processing unit with 12 GB of memory, which is available in the Ilifu computational cluster. Based on the data splitting strategy, a leave-one-out approach was utilized to train the model, and the trained model was validated for 50 epochs on the training and validation data, respectively. The data was randomly shuffled at the start of each epoch, with performance metrics recorded at the end of each epoch. Model checkpoints were generated, storing the best model weights at the end of each epoch. A summary of the workflow used in this study is shown in Figure 2 and a Nextflow pipeline was created for ease of extension and reproducibility, and is available on GitHub (https://github.com/Rudolph-afk/Zoon0PredV).

A graphical representation summarizing the workflow used in this study. POC, proof of concept.
Model evaluation and proof of concept
A small-scale study consisting of 3 imbalance class distribution scenarios was conducted, and the results obtained were used to examine under which class distributions the developed model achieved the best generalization performance. Three models were trained on these varying proportions derived as follows:
(a) The first dataset represented the complete KW-1160 dataset with no modifications, that is, (278 791 samples for the positive class and 38 770 entries for the negative class).
(b) The second dataset was an under-sampled derivative with the majority class being only two-thirds (67%) greater than the minority class (57 862 entries for the positive class and 38 768 samples for the negative class).
(c) The final under-sampled derivative dataset represented equal proportions of the majority and minority classes, containing 38 768 samples for each.
The model was evaluated using the test data, and the model performance on accuracy, precision, recall, F1 score, area under the receiver operating characteristic curve (ROC-AUC), and the Matthews correlation coefficient (MCC). Accuracy is the ratio between the correctly classified samples and the total number of samples in the test dataset. 68 It is given as

Precision is calculated as the ratio between correctly classified samples and all samples assigned to that class. 68 It is computed as

The recall, also known as the True Positive Rate or Sensitivity, is calculated as the ratio between correctly classified positive samples and all samples assigned to the positive class. 69 It is given as

The F1 score is the harmonic mean of precision and recall, 70 and is calculated as

MCC measures the correlation between the true and predicted classes. 71 It is computed as

where TP denotes true positive, FP is false positive, FN is false negative, and TN denotes true negative. For accuracy, precision, recall, and F1-score, the ideal model achieves a value of 1.0, while the poorest performance corresponds to 0. For MCC, the range of predictive value is from −1 (total disagreement) to +1 (perfect prediction), with 0 meaning random guessing.
The performance of the model was evaluated using a test dataset of 19 326 protein sequences, comprising 11 398 positive and 7928 negative samples. To provide a comprehensive assessment of the model’s performance, a confusion matrix was employed, where other performance metrics were also computed. Furthermore, the model was used for proof-of-concept prediction using the proof-of-concept (POC) samples.
Results
The goal of this study is to develop a CNN model for the prediction of potentially zoonotic organisms, using viral surface proteins that facilitate host cell entry as input data. This section presents the results of the empirical investigation conducted in the study.
Model performance on imbalance classes
Table 3 presents the model results for the selected evaluation metrics for the 3 imbalanced class distributions, under which the best model-skewed class condition is determined. The results in Table 2 show that the model demonstrated distinct performance patterns across the 3 skewed class datasets, which reflect the effect of class distribution on predictive capacity. On the full imbalanced dataset (ZoonosisFull), the model achieved high overall accuracy (93.57%) and a strong ROC-AUC score (0.97), indicating that the model successfully captured important sequence and image-level patterns in the dataset. However, the imbalance led to reduced sensitivity to the minority class, with 5575 false negatives recorded.
Evaluation metrics for the validation dataset with 3 imbalanced class distributions.

As indicated by Chicco and Jurman, 72 when a model achieves a high predictive accuracy and is accompanied with a low MCC, the resultant effect is the sensitivity of the model to imbalanced classes in a dataset, which is observed in the ZoonosisFull dataset. In addition, while the F1 score (0.96) indicates that the model maintained a balance between precision and recall, the lower MCC of 0.74 highlights its weaker ability to generalize across classes when exposed to disproportionate class sizes. For zoonotic surveillance, this imbalance implies that a substantial number of zoonotic samples may potentially remain undetected.
The two-thirds under-sampled dataset (ZoonosisTwoThirds) offered a better trade-off between accuracy (95.37%), sensitivity, and specificity. False negatives were markedly reduced (844), while false positives remained relatively low (775). These improvements were reflected in the higher MCC (0.90) and ROC-AUC (0.99), which indicate that the reduction of skew allowed the model to more effectively capture discriminative features across both classes, resulting in improved confidence in model predictions.
The one-to-one balanced dataset (ZoonosisOne2One) provided the strongest overall performance, with the highest accuracy (97.14%), F1 score (0.97), MCC (0.94), and a ROC-AUC of 1.00. Both false positives (129) and false negatives (671) were minimized, demonstrating that the model generalized effectively without favoring one class over the other. This outcome illustrates that the balanced training dataset allows the network to fully capitalize on its representational power to generate reliable and interpretable predictions. Importantly, this improved model performance highlights the role of balanced class distribution in shaping the predictive integrity of the model across both classes.
Biological insight into FCGR image classification
Frequency Chaos Game Representation, employed in this study, generated greyscale images of 224x224 pixels (examples of the generated features for 4 entries are shown in Figure 3. The FCGR image is a large icosagon, which contains 20 edges and 20 icosagons, 73 with the edges representing each of the 20 standard amino acids, translated from nucleic acid sequences in the protein database.50,74

The frequency chaos game representation (FCGR) of 4 virus surface proteins: (A) influenza B virus nucleoprotein (560aa), (B) human orthopneumovirus major surface glycoprotein G (315aa), (C) simian immunodeficiency virus envelope glycoprotein gp160 (865aa), and (D) influenza A virus hemagglutinin (566aa).
Protein sequences derived from methods such as chromatography, mass spectrometry, and X-ray crystallography 75 may occasionally result in errors whereby an amino acid is not clearly identified.75-77 For example, a precise distinction between aspartic acid or asparagine, or glutamic acid or glutamine may result in the presence of different letter representations—B (aspartic acid [A] or asparagine [N]), J (leucine [U] or isoleucine [W]), X (unknown amino acid), Z (glutamic acid [G] or glutamine [Q])—in the sequenced proteins. This is an important consideration when using FCGR, as it only recognizes the standard 20 amino acids and it is assumed that these different letter representations are omitted in the FCGR software.
In addition, 2 recently discovered amino acids are considered part of the proteinogenic code; selenocysteine, and pyrrolysine, represented by the letters U and O, respectively. 78 It is not clear how FCGR would deal with these amino acids should they occur in a given protein sequence, and while this was not of concern in this study, it is an important consideration when FCGR is applied to studies involving proteins containing these unique amino acids.
Model test results
The model performance on the test data, evaluated across 50 epochs, is shown in Figure 4.

The model performance during training and validation on each epoch: (A) training and validation accuracy through the iterative model training and (B) training and validation error/loss through the iterative model training.
The training and validation accuracy of the model exhibited 3 distinct phases over the course of training. During the initial phase (epochs 0-8), both training and validation accuracy remained around 60%, indicating that the network was in the early stages of learning and had not yet fully captured discriminative image features from the dataset. In the transition phase (epochs 8-12), an increase in accuracy was observed for both sets, reaching approximately 95% to 100%. This rapid improvement indicates that the model successfully identified key patterns in the dataset necessary for accurate virus host classification. During the stabilization phase (epochs 12-50), accuracy for both training and validation converged near 96.8% (the best model was obtained on the 48th epoch, which achieved 96.80% accuracy and 0.92 MCC on validation data) and remained stable, demonstrating effective generalization and minimal overfitting. The near-overlap of the training and validation accuracy curves throughout the training process reflects a robust model that performs consistently across both datasets.
Furthermore, the loss curves are similar to the trend observed in accuracy, which illustrates the effectiveness of the weight optimization process during model development. In the initial phase (epochs 0-8), training loss was high (~2.0) while the validation loss was recorded around 0.6, indicating poor early and unstable performance. During the transition phase (epochs 8-12), loss values decreased steadily for both training and validation sets, corresponding to an increase in model accuracy. In the stabilization phase (epochs 12-50), training and validation losses converged to very low values (0.3-0.4) and remained nearly identical, indicating that the model continued to optimize effectively while maintaining strong generalization. The close alignment of the loss curves further confirms that the model avoided underfitting or overfitting.
The performance of the model at predicting the minority (ie, positive class) is also presented in Figure 5.

Confusion matrix illustrating the performance of the model with 11 245 true positives, 7469 true negatives, 259 false positives, and 353 false negatives.
The analysis of binary classification metrics from the confusion matrix in Figure 5 reveals a robust performance of the model across various indicators, with accuracy at 96.83%, precision at 97.75%, recall at 96.96%, F1 score at 97.35%, and specificity at 96.65%. The high accuracy signifies that the model correctly classifies approximately 97% of all samples, which illustrates solid overall performance despite class imbalance in the test dataset. Notably, the precision rate indicates that when the model predicts a positive sample, it is accurate 97.75% of the time, indicating a low false positive rate. Similarly, the high recall shows the capability of the model to identify 96.96% of actual positive samples, emphasizing the robustness of the model in critical scenarios where missing positive samples could have serious implications. 79
Furthermore, while the model achieved high precision and recall, the sensitivity of these results in model evaluation warrants consideration. Precision focuses on minimizing false positives, while recall aims to capture all true positives. For imbalanced datasets, the F1 score and specificity together provide a comprehensive evaluation of the performance of a classifier across both positive and negative samples. 72 Although the model demonstrates excellent performance, there remains room for improvement, particularly in addressing the current counts of false positives and false negatives, which are 259 and 353, respectively. Furthermore, the model achieved a MCC of 0.93, which, as noted by Chicco and Jurman, 72 is a more reliable metric than both the F1 score and accuracy for evaluating binary classification performance. In addition, the ROC-AUC score of the model was 0.99, illustrating its robustness and exceptional performance on the test dataset.
For proof-of-concept testing, 4 entries from A0A1W5YKT3 (Bat coronavirus, Spike glycoprotein), A0A0P0KH07 (Human coronavirus 229E, Spike glycoprotein), Q5EED8 (Human immunodeficiency virus 1, Envelope glycoprotein), and A0A0M4Q8U3 (Influenza D virus, Nucleoprotein) were tested on the model. The Bat coronavirus and Influenza D virus were correctly predicted as non-human infecting viruses with probability scores of 0.0010 and 0.00042, respectively, which are below a selected threshold of 0.5 in the study. The significantly low probability scores indicate that the proteins do not have signatures associated with sequences from viruses which have been reported to infect humans. The scores also indicate that these viruses potentially require substantial sequence evolution to permit future species barrier cross-over. The Bat coronavirus is indicated to have 101 hosts in the KW-1160 dataset, and the low probability scores obtained from the model, coupled with the wide host range of this virus, illustrate the complexity and rareness of zoonotic events, thus possibly supporting the pinhole model. 3 However, the selected entry for the proof-of-concept may be a strain which has not undergone mutations to allow species crossover.
Discussion
Several epidemics and pandemics are linked to host switching by viral pathogens, originally established in an animal host or reservoir. 80 Epizootic and zoonotic diseases are driven by spill-over of a pathogen to a previously unexposed, non-susceptible host, and when these events occur, the resultant outbreaks can have devastating consequences. 80 Despite the clear threats to public health and biosecurity which are caused by the emergence and re-emergence of zoonotic diseases, many host crossover events are not detected or reported, and the modeling of infectious disease to predict spill-over remains constrained by several challenges.81,82 Public health research priorities toward emerging infectious diseases are largely focused on the detection and surveillance of EIDs, as well as the identification of factors driving transmission, to intervene for public safety and mitigate the effects of disease.7,83 Detection efforts are focused on deployment of analytical, laboratory-based methods for identification of microorganisms, ranging from traditional culturing to modern molecular and “-omics” techniques.7,83,84 In addition to traditional surveillance efforts, various statistical and ML models, which make use of different features and prediction targets, have been developed to predict cross-species spill-over of novel and re-emerging infectious agents, as well as transmission dynamics once an outbreak has occurred.15,16,85
In this study, a machine learning approach was used to develop a model that predicts the zoonotic potential of pathogenic species by learning protein sequence patterns of viral pathogens. Considering that host specificity is critically dependent on viral interaction with host cells, receptor binding (and changes thereof) inevitably plays a vital role, 80 and as such, viral proteins involved in pathways of host cell entry were used to train, validate, and evaluate the model. From the dataset perspective, the positive samples used in this study consisted of viral pathogens known to infect human hosts, while those documented not to have a human host formed the negative samples. The trained model could then be used to predict if an unknown virus would be capable of infecting a human host cell, based on the consistency of protein sequence patterns learned during model training.
The Chaos Game Representation (CGR) is a sequence representation scheme inspired by chaos theory in physics, originally proposed by Jeffrey 59 as a visual representation scheme for DNA sequences. Frequency Chaos Game Representation (FCGR) is an adaptation of the original CGR method and has been modified to accommodate protein sequences. 73 Another variant of CGR proposed by Mu et al, 86 called DCGR, incorporates amino acid physiochemical attributes, which are important determinants of protein structure, interaction, and function. Several encoding methods are available which use mathematical transformations as well as pre-computed embeddings,87-91 however, FCGR is an underrepresented feature encoding method, shown to achieve good metrics in our study.
Convolutional neural networks were used in this study due to their exceptional image classification capability, particularly for the FCGR images. There is no standard convention for building CNN models due to varying performances of different model architectures. 92 This is often further complicated by the presence of a multitude of parameters which require tuning93,94 and can include the number of layers to use, the number of nodes within each layer, a specific optimizer, the learning rate of the optimizer, the activation function, and others. 94
The high accuracy obtained with the developed CNN model in this study shows the excellent capability of convolutional neural networks, coupled with the FCGR features.
It is noteworthy to mention the hyperparameter optimization approach resulted in significant benefits during the CNN model development. The model provided optimal performance with a single layer and multiple nodes within its architecture due to the characteristics and complexity of the dataset used in this study. Based on the complexity of the dataset, with reference to large sample sizes, the model reached faster convergence and utilized lower computational time during training, which highlights the stability of the model in achieving high performance across the selected metrics.
Furthermore, previous models often focus on virus-host interactions 15 and analysis of host receptor similarity,23,25 which tend to limit the utility of the latter model, particularly if a virus emerges and uses a different host receptor to those already known. However, the developed model achieved high performance because of the significantly large dataset used in this study (since the model is exposed to a sufficient number of samples for efficient training and better generalization), when compared with the quantity of data used in previous studies, such as the 10 host receptor protein sequences in Bae and Son, 25 211 interaction pairs in Yan et al, 24 and 277 host receptor protein sequences in Cho and Son. 23 In addition, the training data used in this study consist of a highly diverse dataset, which included viruses reported to infect plants as well as those reported to infect non-eukaryote organisms.
Also, CGR has been applied to viral proteins, with tools such as PhaVIP, 95 which classifies phage virion proteins, and Spike2CGR, which models coronavirus spike proteins. 96 These approaches convert sequences into images and employ convolutional neural networks for classification tasks, which aligns with our approach. In a related context, deep learning frameworks such as VIDHOP, tested on influenza A virus, rabies lyssavirus and rotavirus A, achieving AUC of between 0.93 and 0.98 for each viral species, 36 HostNet, 42 and Virus2Vec, tested on real-world coronavirus spike and rabies virus sequence data, 97 employ embedding strategies or neural architectures to classify viral hosts with high accuracy. Compared with these embedding-driven models, our FCGR approach provides a holistic frequency representation that does not rely on motif or k-mer context but instead emphasizes global compositional structure.
From protein language modeling (pLMs), recent algorithmic advances such as ESM2 and related frameworks have demonstrated remarkable performance in tasks such as host tropism prediction, escape mutant detection, and structural inference.88,98 Further advances in protein and genomic language modeling demonstrate the capacity of sequence-only learning approaches to capture biologically meaningful signals related to viral evolution and pathogenicity. Virus specific generative models such as SpikeGPT299 and SARITA 100 have shown that large language models (LLMs), not only generate realistic SARS-CoV-2 spike protein sequences, but can also retrospectively anticipate the emergence of mutations associated with altered transmissibility, and be used to examine pathogen evolution.99,100 At a broader scale, models such as Evo 2, trained on DNA and RNA sequences spanning all domains of life, can enable accurate prediction of mutational effects and pathogenicity. 101
While these models highlight the power of large-scale language modeling, they are primarily designed as generalist or pathogen-specific and are focused on variant effect prediction and evolutionary forecasting, rather than direct zoonotic risk assessment. Large language models and pLMs generally require substantial computational resources, however, our study offers a computationally efficient alternative tool for early detection of host switching in emerging viruses, through a targeted, protein sequence approach which focuses on viral surface proteins involved in host cell entry.
Interestingly, 5 phage portal protein (PP) samples from bacterial and plant hosts were observed in the false positive predictions, namely, A0A0K2FHA1 (Achromobacter phage phiAxp-2), A7TWJ1 (Staphylococcus virus tp310-2), I7HHN4 (Helicobacter virus KHP30), I7KR94 (Yersinia virus R1RT), and M4QNQ7 (Tetraselmis viridis virus S20). Portal proteins have a low sequence similarity but highly conserved functionality, playing a role in bidirectional viral DNA passage. 102 These phage portal proteins are being considered as potential antiviral drug targets in herpes simplex virus infections. 103 The “plasticity” of phage PP may explain the erroneous classification by the model, due to the presence of signatures consistent with proteins involved in viral entry into human host cells. This observation may indeed be of interest for further investigation, as false positives in this dataset may contain samples which could be considered for therapeutic experimentation, as in Dedeo et al. 103 The other false positives may be as a result of protein similarity. However, this does not eliminate the possibility that some of the false positives may be of future concern, having the capability to bind to human host cells, but still lacking machinery for sustained infection and replication.
A surprising observation in the false negative class was the erroneous classification of 31 Human Immunodeficiency Virus (HIV) entries. This was unexpected, as HIV is an established, long-term endemic virus with characteristic signatures of viruses with reported human hosts. Investigation of some of the HIV samples, such as A0A2P1DQ38, Q7SPP5, and A0A2P1DR91 showed the warning “Lacks conserved residue(s) required for the propagation of feature annotation,” according to UniProtKB. Computationally derived feature annotation is reliant on existing knowledge and annotations based on sequence homology, resulting in errors which are propagated in databases and give rise to contradictory interpretations of the data.104,105
Thus, we demonstrated the capability of generating a robust model with good performance metrics. The insights generated from the developed model indicate the existence of patterns in the sequences of virus surface proteins that interact with host cells at the initial stage of infection. The insights may also be indicative of zoonotic potential, and it could possibly aid in identifying zoonotic viruses, using sequence data extracted from pathogen surveillance programs, as input into the model. Taken together, the results from this study showed the presence of consistent patterns in surface proteins of viruses reported to infect humans which differ from surface proteins of viruses which do not infect humans. From a biological perspective, this is expected, as host range is determined by successful infection,17,106,107 and virus-host cellular protein-protein interactions are a key mechanism.27,108,109 Furthermore, it is plausible that a similar approach could be adopted to design a model which predicts epizootic events for hosts other than humans, and particularly for animals of domestic and agricultural importance. In addition, the approach is flexible enough to support multi-category classification, with a simple modification of the final layer in the model architecture, such that a single model could potentially predict cross-species likelihood for several hosts rather than for a single host. Such a model would be a valuable application of ML to the One Health initiative, moving the focus from solely humans to other host organisms.
To further validate the effectiveness of the developed model in line with the work done in literature, Table 4 presents a comparison between our Zoon0PredV model and other models.
Accuracy and AUC comparison between Zoon0PredV and other models in literature.

From Table 4, compared with prior approaches, Zoon0PredV demonstrates superior predictive performance. While IILLS 24 and GBM 38 did not report accuracy, their AUC values (0.90 and 0.773, respectively) are notably lower than that of Zoon0PredV (0.99). ViCIPR 23 reported an accuracy of 83.3% with an AUC of 1.00, but its accuracy lags significantly behind the 96.78% achieved by Zoon0PredV. Taken together, these results indicate that our model delivers a more balanced and consistently high performance, combining both excellent accuracy and discriminatory power (AUC). In addition, the Zoon0PredV learned patterns present in viral surface proteins such that even if a new virus emerges, targeting an uncommon host receptor, the viral protein patterns will still be detected. To our knowledge, no previous study identified in our literature search has combined FCGR of viral surface proteins with CNNs to develop a machine learning model aimed at predicting viral species cross-over events.
A number of limitations are acknowledged in this study. Although this study presents promising findings, the model has not been compared with other models that utilize common dataset, and it may contain biases that have not been identified in this analysis. Furthermore, we included limited viral sequences in our POC dataset (n = 4), and while biologically plausible predictions were observed, the inclusion of additional sequences (derived from databases or synthetically generated) would provide larger scale validation of the biological relevance and predictive performance of our model.
Several areas of research are investigating pattern analysis for biological inference, and as such, comparison and ablation studies using classical and advanced sequence embedding tools such as those performed by Lin et al 88 and Jiao et al 91 are needed to understand model performance and to examine if a hybrid approach to the task can yield more optimal results. Inherent data bias is an additional consideration that could potentially affect the model performance and Generalizability. For instance, a specific strain may have the capacity to infect several hosts, but in a database, may appear to infect a single host species. This phenomenon may be due to research priorities, based on perceived host “value” (human vs horse). In this way, even if the virus can infect additional hosts, systemic bias in data representation and data priority exists. 110 It is envisaged that with the increased research in One Health, that research priority will become less skewed.
Conclusions
The rise in epidemic and pandemic events of zoonotic origin has prompted the need to efficiently predict and mitigate future incidents. This study aimed to produce a proof-of-concept approach to predicting the zoonotic potential of viruses. A zoonosis prediction model was created using, as input, FCGR-encoded sequences of virus surface proteins, which facilitate viral entry into host cells. The model developed in this study showed the existence of patterns in the sequences of virus surface proteins that interact with host cells at the initial stage of infection and are indicative of zoonotic potential It should, however, be noted that inferences about host-virus associations using sequence data alone may not capture the biotic and abiotic factors, which play important roles in host tropism. 42 The CNN binary classification model obtained a 96% accuracy on the test data (ie, generalization performance), outperforming other approaches found in literature. However, the approach we used may benefit from using data with clear evidence of zoonosis to produce a more robust model. In addition, the study developed a binary classification model which focuses on cross-species prediction to human hosts, and as such, we suggest that future studies include other host organisms by building a multi-categorical model representing the varying host species, in line with a holistic One Health approach. The use of natural language processing tools such as ESM2, the largest language model trained to date for a variety of protein-related tasks, could also be used with our specific dataset to further refine our methodology and strengthen the current model, thereby increasing the reliability and subsequent use of ML technologies in public health-related research and pathogen surveillance.
Future research should incorporate systematic benchmarking across different encoding methods to evaluate their impact on zoonosis prediction performance. The study will also be extended to include more recent deep learning architectures, such as ResNet, 111 EfficientNet, 112 and Vision Transformers (ViTs), 113 to assess their relative effectiveness in molecular zoonosis prediction. In addition, the dataset, currently based on UniProtKB 2021, will be updated to the latest releases to improve data recency, increase the number of available sequences, and enhance generalizability. These updates will further enable evaluation of the robustness of the model against newly discovered viral sequences. Collectively, these efforts aim to enhance both the predictive accuracy and practical applicability of the proposed approach in real-world scenarios.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322251415123 – Supplemental material for Zoon0PredV: Potential Virus Species Crossover Prediction Using Convolutional Neural Networks and Viral Protein Sequence Patterns
Supplemental material, sj-docx-1-bbi-10.1177_11779322251415123 for Zoon0PredV: Potential Virus Species Crossover Prediction Using Convolutional Neural Networks and Viral Protein Sequence Patterns by Rudolph Abel Serage, Clement Nthambazale Nyirenda, Taiwo Gabriel Omomule, Alan Gilbert Christoffels and Dominique Elizabeth Anderson in Bioinformatics and Biology Insights
Supplemental Material
sj-zip-2-bbi-10.1177_11779322251415123 – Supplemental material for Zoon0PredV: Potential Virus Species Crossover Prediction Using Convolutional Neural Networks and Viral Protein Sequence Patterns
Supplemental material, sj-zip-2-bbi-10.1177_11779322251415123 for Zoon0PredV: Potential Virus Species Crossover Prediction Using Convolutional Neural Networks and Viral Protein Sequence Patterns by Rudolph Abel Serage, Clement Nthambazale Nyirenda, Taiwo Gabriel Omomule, Alan Gilbert Christoffels and Dominique Elizabeth Anderson in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
The authors wish to acknowledge Mr Peter Van Heusden and Dr Nasr Eshibona from the South African National Bioinformatics Institute for code evaluation and guidance.
Ethical Considerations
Ethical approval was not required for this study.
Author Contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this project was supplied by the DSI/NRF Research Chair in Bioinformatics, Grant number 64751.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Use of AI
No scientific data has been generated or modified using AI tools such as ChatGPT.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.