
Abstract
Background
Leishmaniasis is a severe zoonotic disease which significantly impacts public health worldwide. It is caused by protozoan parasites belonging to the genus Leishmania (order Kinetoplastida, family Trypanosomatidae). 1 According to the most recent data published by the World Health Organization, the estimated annual incidence ranges between 700 000 and 1 million new cases, with more than 1 billion people at risk across 98 countries. 2 These data highlight the substantial global health burden of the disease, particularly in tropical and subtropical regions of low- and middle-income countries. 3
This infection presents with a spectrum of clinical manifestations, largely influenced by the infecting Leishmania species and the host immune response. Clinically, it is categorized into 2 principal forms: visceral leishmaniasis (VL) and cutaneous leishmaniasis (CL). 4 Among these, CL represents the most prevalent form, affecting impoverished populations with limited access to healthcare services. Although often self-limiting, CL can lead to permanent disfigurement and significant socioeconomic impact due to reduced productivity. 4
In Brazil, 7 Leishmania species have been identified as etiological agents of CL, underscoring the disease’s complexity and public health relevance in both Brazil and the broader Latin American region. The country reports an average of 21 000 new CL cases annually, corresponding to an incidence rate of 8.6 cases per 100 000 inhabitants over the past 5 years. This epidemiological scenario reflects the endemic nature of CL, with Leishmania (Viannia) braziliensis recognized as the predominant species responsible for most cases. 5
The absence of a licensed human vaccine, combined with the limitations, high costs, and toxicity associated with current therapeutic options, represents a major barrier to the effective prevention, control, and treatment of leishmaniasis. 6 In this context, the increasing use of in silico approaches, such as reverse vaccinology in computational immunology, has emerged as a promising strategy for identifying novel antigens and expediting vaccine candidate selection at reduced time and cost. In a previous study, our research group employed in silico approaches to identify epitope candidates, which were mined from genomic analyses, with the aim of stimulating a cellular response against L (V.) braziliensis in humans. 7
Subsequent computational analyses identified 10 epitope candidates (peptides), selected based on their higher in silico predicted affinities across diverse major histocompatibility complex alleles. Experimental validation through serological assays demonstrated their immunogenic potential, demonstrating their ability to stimulate effector cell proliferation and cytokine production.7,8
The use of synthetic peptides has significantly broadened the scope of scientific research, especially in diagnosing neglected diseases. These peptides are valuable as sources of epitopes and pure molecules for detecting circulating antibodies and antigens. One of their key advantages lies in their cost-effectiveness and ease of large-scale production, which allows for consistent reproduction and precise control over their properties, ensuring their effective application in final diagnostic products. In addition, they offer advantages in terms of storage and transportation, allowing their application across different locations and time points.9,10 This rising interest in synthetic peptides plays a key role in advancing new diagnostic methods for a variety of diseases.11 -15 Although significant progress has been made, there have been limited studies examining the structural interactions between synthetic peptides and specific IgG subclasses, creating a gap that hinders advancements in diagnostic precision. Gaining a deeper understanding of these subclass-specific interactions is crucial for improving peptide selection and increasing the accuracy and reliability of immunoassays.
Alongside the ongoing advancements in diagnostic technologies, there is still a strong need for better tools that can accurately assess the immune response triggered by Leishmania infection. The immune response to Leishmania infection is highly complex, involving a range of cellular components. In the context of CL, this response is notably characterized by the production of IgG antibodies, particularly the IgG1 and IgG3 subclasses. Recognizing the critical role of IgG in the host immune response to L braziliensis, this study aims to predict (in silico) and experimentally evaluate (in vitro) the binding affinities of our group previously validated immunogenic peptides7,8 across different IgG isotypes. Through the characterization of peptide-IgG subclass interactions, the research aims to advance the understanding of the humoral immune response in CL, where the antibody profile may correlate with both parasite burden and disease chronicity. To our knowledge, this study represents the first comprehensive in silico docking analysis of 10 peptides against 124 antibody structures (including several IgG subtypes), aiming to characterize antigen-antibody (Ag-Ab) interaction and correlating these findings with in vitro experimental data.
Methodology
Ethical aspects
This study was reviewed and approved by the Ethics Committee in Human Research at the Aggeu Magalhães Institute (Approval code: 11083812.7.0000.5190). Written informed consent was obtained from all participants prior to their inclusion in the study.
Patients and samples
The study included samples from individuals of both genders, aged 18 years and older. Participants were categorized into 2 groups: those positive for CL (N = 50), confirmed by positive results in both polymerase chain reaction (PCR) and direct examination tests, and healthy controls (N = 28), who tested negative in both tests. The inclusion criteria for patients followed the standards established by the Reference Service of the Aggeu Magalhães Institute (Oswaldo Cruz Foundation—FIOCRUZ) and the Dermatology Clinic of the Federal University of Pernambuco (UFPE), requiring the presence of active lesions, no prior treatment for leishmaniasis, and a positive diagnosis confirmed by 2 distinct methods (molecular/PCR and parasitological/ direct microscopic examination of lesion smears). Exclusion criteria included absence of lesions, presence of other dermatological disorders, and previous leishmaniasis treatment.
Peptide preparation
The immunogenic peptides used as antigen candidates in this study were derived from the proteome of L (V.) braziliensis. These peptides were selected and validated through previous studies conducted by our research group, aiming to identify epitope candidates in a reverse vaccinology approach for L (V.) braziliensis.7,8 Eight peptides originated from 3 distinct hypothetical proteins: peptides I, IV, V, and VIII were derived from LbrM.34.3630; peptides VI, VII, and X from LbrM.01.0110, a protein conserved across Trypanosoma and Leishmania species with currently undefined properties and functions; peptide III from LbrM.06.0820, a hypothetical protein unique to Leishmania and closely related taxa, absent in Trypanosoma species; and peptides II and IX from LbrM.14.1680, annotated as a putative synaptojanin, an inositol/phosphatidylinositol phosphatase implicated in vesicle-mediated transport. The primary sequences of these peptides are presented in Table 1. The peptides were analyzed individually and also grouped into 2 pools. Pool 1 (comprising peptides I, III, V, VI, and VII) exhibited a specific response profile, stimulating groups of patients with CL, while pool 2 (composed of peptides II, IV, VIII, IX, and X) showed a nonspecific response profile, stimulating both the tegumentar leishmaniasis (LT) patient group and the healthy control group. All peptides were commercially synthesized by Genome Biotechnology (Brazil) and purified by high-performance liquid chromatography (HPLC) to achieve a final purity greater than 95%. Each synthetic peptide was then stored at −20°C until further use.
Peptide sequences and source protein identifiers of each pool.

Legend: Pep = Peptide.
Selection of antibody structures
Immunoglobulin G (IgG) structures were retrieved from the SabDab structural antibody database (opig.stats.ox.ac.uk/webapps/newsabdab/sabdab), which compiles all antibody structures from the Protein Data Bank (PDB) and structure selection based on attributes. For this study, the following selection attributes were used: (1) “All” for “Antibody type”; (2) “All” for “Experimental method”; (3) “Homo sapiens” for “Species”; (4) no specific values were used for “Resolution cutoff” and “R-factor cutoff”; (5) “Peptide” for “Antigen type”; (6) “All” for “Light chain type”; (7) “All” for “Has constant region”; (8) “All” for “Has affinity value”; and (9) no data were specified for “Residue at Chothia position..” It is important to note that this search for antibody structures was conducted in September 2020, and therefore, the attributes “Is in CoVAbDab,” “Is in TheraSabDab,” and “Keyword query” were not available at that time and, hence, these setups were not used. The initial search yielded 411 antibody structures; however, many were duplicates or incomplete, lacking either the light or heavy chain. In this context, a screening strategy has been employed in order to remove the aforementioned duplicate and missing chain structures, using the following exclusion and tie-breaking criteria: (1) presence of a co-crystallized peptide approximately 15 amino acids in length; (2) highest available resolution (lowest R-factor); (3) most recent publication date; and (4) presence of both light and heavy chains. After applying these criteria, a total of 124 unique and curated IgG structures were selected for further analysis (see supplementary material).
Modeling antigen-antibody (Ag-Ab) complex interaction
Prior to conducting docking calculations, all antibody structures underwent curation, where water molecules and chains unrelated to the light (L) and heavy (H) chains were removed. In addition, hydrogen atoms were added to complete the valence of all atoms in the biomacromolecules, using AutoDock Tools. 16 The antibody structures were also renumbered to the Chothia numbering scheme, using the Abnum antibody numbering server (www.bioinf.org.uk/abs/abnum), selecting the “Number your structure” option. To generate new antibody-epitope complexes (antibody + epitope candidate), the FixBB and Relax protocols from Rosetta’s framework 17 were employed. The FixBB protocol replaced the amino acid sequence of the co-crystalized peptide for the amino acid sequence of the proposed peptides, generating 10 Ab-Ag complexes for each one of the 124 antibodies, whereas the Relax protocol allowed only the replaced peptide to adapt into the more stable geometry (local optimization), only constraining the antibody’s structure. Before applying the FixBB and Relax protocols, the co-crystalized peptides with lengths different than 15 amino acids were adjusted using PyMol software (pymol.org), by removing the surplus or adding alanines. Ultimately, a total of 1240 (10 × 124) Ag-Ab complexes were generated for docking analyses.
Docking calculations
The SnugDock protocol, 18 available within the Rosetta framework, 19 was employed to perform the docking calculations for the 1240 Ag-Ab complexes. This tool simulates induced fit for the Ag-Ab interaction, allowing flexibility of the CDR loops, side chains, and VL-VH orientation minimization. All complexes were subjected to the standard parameters and recommended conditions for SnugDock, as outlined in the Rosetta Commons documentation (docs.rosettacommons.org/docs/latest/application_documentation/antibody/snugdock).
This approach uses an accurate and optimized setup for docking simulations of antigen-antibody complexes, enhancing the reliability of the results. Our research group developed a Python script capable of preparing the necessary files for docking calculations for all the 1240 Ag-Ab complexes used in this study. After the calculations, a database on the MySQL 8.0 platform was constructed using another Python script also developed for this purpose, containing all descriptors resulting from each docking solution. Finally, the docking results were compiled into a summary dataset, generating a spreadsheet that included the best score, mean, and standard deviation for each descriptor stored in the database.
Intermolecular interaction analysis
The BINANA program 20 was employed for the intermolecular interaction analysis, choosing its default setup, except for the hydrogen bonding distance, which was altered to a maximum of 3.5Å. All files, in PDBQT format, were obtained by using AutoDock Tools. 16 The images were generated using the Pymol program (pymol.org). 21
Enzyme-linked immunosorbent assay using synthetic peptides of L (V.) braziliensis
The enzyme-linked immunosorbent assay (ELISA) assay was conducted following the protocol previously established by the Laboratory of Immunopathology and Molecular Biology at the Aggeu Magalhães Institute (IAM). Each peptide was individually coated onto the plate, and concentrations of 100, 200, 400, and 600 ng were tested for assay optimization. The plates were incubated at 4°C for 18 hours in a humidified chamber. After incubation, the plate was washed 3 times with phosphate-buffered saline (PBS) 1X (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.4) containing 0.05% Tween 20% and 5% bovine serum albumin (BSA). Serum samples were diluted in PBS 0.015 M (pH 7.2) containing 0.05% Tween 20% and 5% BSA. For each antigen, the optimal serum dilution was previously standardized by testing 4 serial dilutions (1:450, 1:900, 1:1800, and 1:3600), with the selected dilution subsequently used for sample evaluation. Positive (serum from CL-positive patients) and negative (serum from healthy individuals) controls were included on each plate, following the same dilution conditions. A volume of 100 µL of diluted serum was added to each well, and the plates were incubated for 1 hour at 37°C in a humid chamber. After incubation, the plates were washed as described previously. Detection was performed using 100 µL of peroxidase-conjugated human anti-IgG (γ-chain specific, Calbiochem) at an optimized dilution of 1:2500, followed by a 1-hour incubation at 37°C. Subsequently, plates were washed 3 times with PBS-Tween-BSA buffer, and 150 µL of chromogenic substrate (5-amino salicylic acid or OPD) containing hydrogen peroxide was added per well. After a 30-minute incubation at room temperature in the dark, the reaction was stopped by adding 100 µL of 1 M sulfuric acid per well. Absorbance was measured at 490 nm using a spectrophotometer. All samples were tested in triplicate, and results were expressed as optical density (OD) values.
Statistical analyses
For the experimental assays, ELISA OD was measured using serum samples from 50 confirmed patients with CL and 28 well-defined healthy controls. Negative controls were rigorously selected from individuals lacking clinical signs of CL and testing negative by both PCR and parasitological methods, thereby minimizing false-positive and false-negative results. Positive controls comprised sera from patients exhibiting active CL lesions. Each sample was analyzed in triplicate on the same plate to ensure assay reproducibility. The mean OD for each sample was calculated, and background correction was performed by subtracting the average OD of the blank control wells. To determine the optimal diagnostic threshold for each peptide, receiver operating characteristic (ROC) curves were constructed using GraphPad Prism software (version 7.0; GraphPad Prism Inc., San Diego, California), and the area under the curve (AUC) 22 was calculated to determine the overall diagnostic performance of each peptide (see Supplementary Materials). The optimal cut-off points were established based on the ROC analysis. Subsequently, sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were calculated. Exact 95% binomial confidence intervals for these parameters were determined using 2 × 2 contingency tables in OpenEpi software (version 3.01; Centers for Disease Control and Prevention, Atlanta, Georgia). All analyses were performed in comparison with the reference standard diagnostic methods employed for case definition in the study population, allowing for a comprehensive evaluation of each peptide’s diagnostic potential for CL. For the in silico data, additional statistical analyses were conducted using PAST software (version 4.12b) 23 and Orange software (version 3.32), 24 focusing on descriptor distribution, clustering patterns, and predictive model evaluation. Correlation analyses were performed using Pearson correlation coefficient to assess linear relationships between variables, with P values <.05 considered significant.
Results
Modeling antigen-antibody (Ag-Ab complex) interaction
A total of 1,240 antigen-antibody (Ag-Ab) complexes were generated by structurally combining 124 distinct IgG structures with the 10 previously selected epitopes from L braziliensis. These complexes were subjected to docking calculations using the SnugDock protocol within the Rosetta framework, employing the recommended parameters, which included the generation of 1000 docking solutions per complex. This process was applied to each of the 1240 Ag-Ac complexes, resulting in an overall number of 1 240 000 solutions, each with its respective structure and docking score. The docking performance was evaluated based on Rosetta Energy Units (REU), with scores predominantly presenting negative values. Notably, more negative interface scores (I_sc) indicate higher predicted binding affinities between the antigen and antibody. The distribution of SnugDock-derived interface scores for all complexes is summarized in Table 2 and illustrated in Figure 1.
Summary of statistical analysis of SnugDock docking score values for 1240 Ag-Ac complexes.
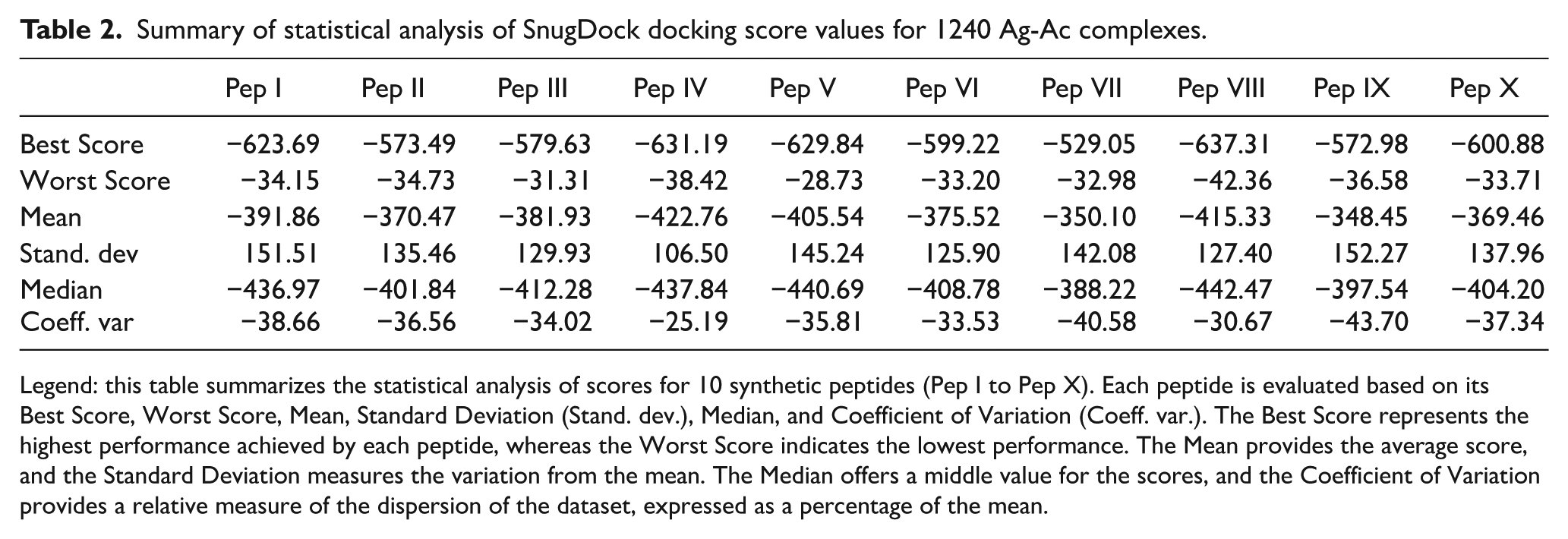
Legend: this table summarizes the statistical analysis of scores for 10 synthetic peptides (Pep I to Pep X). Each peptide is evaluated based on its Best Score, Worst Score, Mean, Standard Deviation (Stand. dev.), Median, and Coefficient of Variation (Coeff. var.). The Best Score represents the highest performance achieved by each peptide, whereas the Worst Score indicates the lowest performance. The Mean provides the average score, and the Standard Deviation measures the variation from the mean. The Median offers a middle value for the scores, and the Coefficient of Variation provides a relative measure of the dispersion of the dataset, expressed as a percentage of the mean.

A box-plot chart showing the distribution of SnugDock docking score values, measured in REU, for the best result of each of the 1240 Ag-Ac complexes.
The best scores range from −529.05 (Pep VII) to −637.31 (Pep VIII), whereas the worst scores range from −28.73 (Pep V) to −42.36 (Pep VIII). The scores for Pep IX exhibited the greatest variability, with a coefficient of variation (CV) of −43.7 REU and a standard deviation of 152.27 REU, indicating a high level of dispersion and inconsistent performance across different antibodies. Pep VII showed a similar pattern, with a standard deviation of 142.08 REU and a CV of 40.58 REU, reflecting significant variability as well. In contrast, Pep IV and Pep VIII displayed greater stability, with closely aligned mean and median values that were highly negative, suggesting both strong and consistent predicted binding affinities across all antibodies evaluated. The symmetrical distribution of scores for these peptides further supports their reliability in docking performance. Notably, the best scores observed for Pep IV (−631.19 REU) and Pep VIII (−637.31 REU) reinforce their favorable and consistent interaction profiles.
Pep I and Pep III, however, presented a degree of asymmetry between their mean and median scores. The more negative median values suggest favorable predicted binding affinities across most antibody-peptide interactions. However, instances where the mean values deviate substantially from the corresponding medians indicate the presence of outlier docking scores, reflecting markedly poorer predicted interactions in a subset of antibodies. These findings indicate that some peptides exhibited a limited number of particularly unfavorable interactions, which disproportionately increased the mean docking scores. This suggests that, although these peptides demonstrated strong predicted binding with certain antibodies, their performance was inconsistent across the full antibody panel. Such variability may restrict their suitability for broad-spectrum diagnostic applications. This asymmetry highlights the need for further evaluation of these peptides to understand their inconsistent performance across different antibodies. Overall, these findings underscore the importance of considering both central tendency and dispersion measures to gain a more nuanced understanding of peptide performance in immunological studies.
The SnugDock protocol provides a list of 45 descriptors for each docking solution. A full list of descriptors can be found at the supplementary material, including several detailed analyses using those descriptors. Figure 2 presents a heatmap of SnugDock descriptors, which offers a detailed view of the interaction profiles for Ag-Ab complexes. The analysis specifically highlights the “Best” result (the most negative interface score, I_sc) and the “Worst” result (the least negative I_sc) for each peptide across the 124 evaluated antibodies. This classification underscores the variability in predicted binding efficiency, illustrating the contrast between the strongest and weakest interactions within the antibody panel.

Heatmap of SnugDock descriptors for Ag-Ab complexes.
The columns correspond to various SnugDock descriptors, including “dSASA_hphobic,” “hbond_sc,” “fa_atr,” and “hbond_E_env,” which quantify key interaction features such as hydrophobic surface area, side-chain hydrogen bonding, attractive van der Waals forces, and electrostatic energy contributions, respectively. The color gradient ranges from blue to red, where blue represents negative values and red represents positive values. For instance, the “dSASA_int” descriptor exhibits prominently positive values on the heatmap. This descriptor is vital for evaluating how much of the surface becomes accessible to the solvent, providing insight into the extent and strength of the interaction between the molecules. Further details are illustrated in Figure 3.
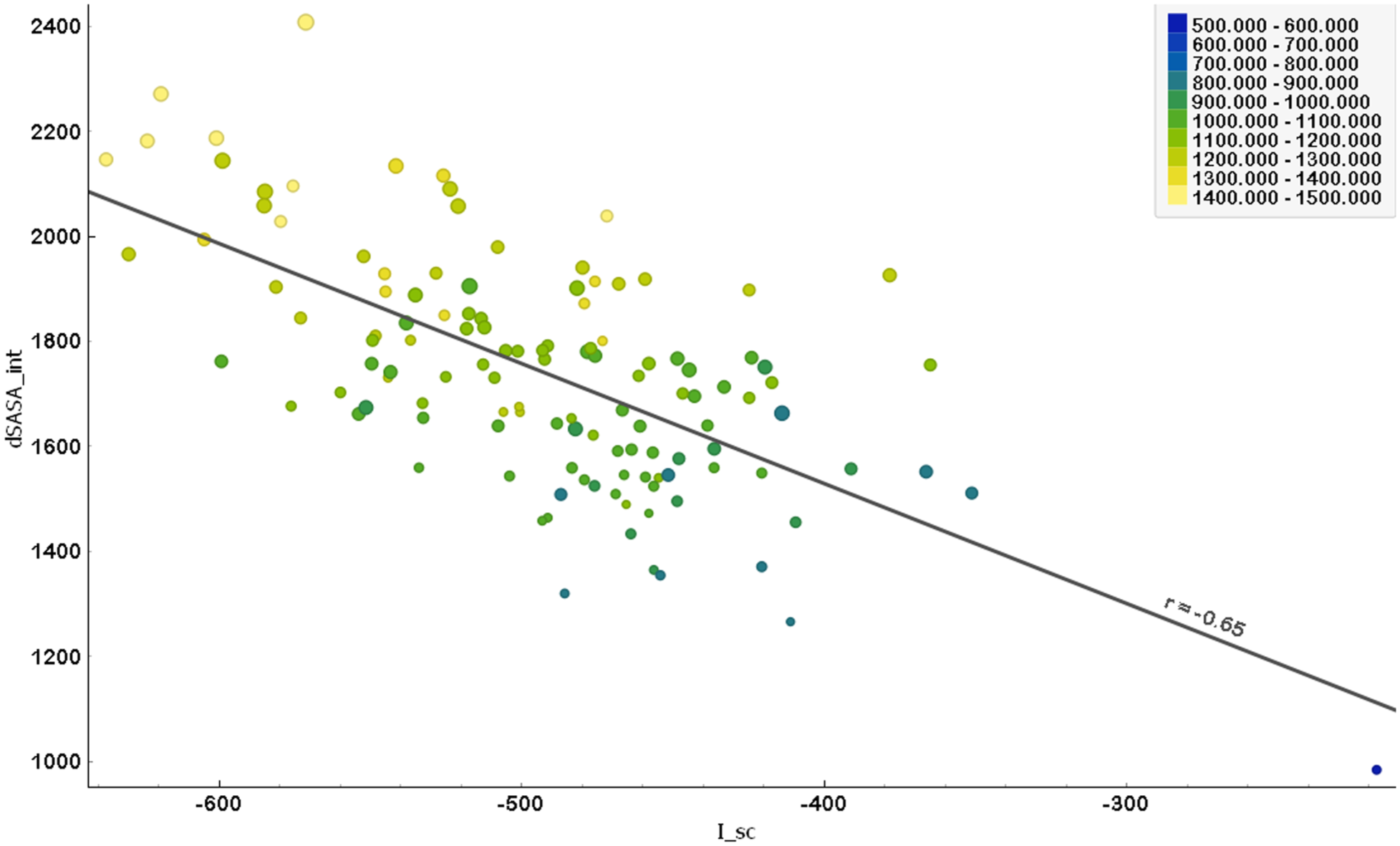
Correlation between interface score (I_sc) and solvent-accessible surface area (dSASA_int).
Figure 3 illustrates the relationship between the interface score (I_sc) and the solvent-accessible surface area (dSASA_int) for Ag-Ab complexes. The x-axis represents the interface score (I_sc), the primary metric used to evaluate binding affinity, whereas the y-axis shows the dSASA_int, encompassing both polar and hydrophobic surface areas. The Pearson correlation coefficient (r = −.652, P < .0001) reveals a moderate negative correlation between I_sc and dSASA_int. This suggests that as the solvent-accessible surface area increases, the interface score becomes more negative, indicating improved predicted binding efficiency. Thus, higher dSASA_int values are associated with more favorable (more negative) I_sc values, reinforcing the significance of surface accessibility for complex stability and interaction performance.
In Figure 3, the color gradient of the data points—from blue to green to yellow—represents the hydrophobic solvent-accessible surface area (dSASA_hphobic). Points that are more yellow indicate larger hydrophobic areas, which correspond to more negative (and thus better) interface scores (I_sc). This suggests that increased hydrophobic surface area enhances interactions at the antigen-antibody interface, resulting in improved binding affinity. In addition, the size of each point represents the polar solvent-accessible surface area (dSASA_polar). Larger points represent larger polar areas, which are also typically associated with more negative (better) I_sc values. This implies that a more extensive polar interface similarly contributes to better interaction at the Ag-Ab interface. Overall, both a larger hydrophobic and polar surface area appear to be beneficial for enhancing the predicted binding affinity for Ag-Ab complexes.
Intermolecular interaction analysis
For each peptide, the intermolecular interactions were mapped using the top-ranked antibody complexes, those exhibiting the highest predicted affinities within the evaluated set. The primary goal of this mapping was to understand the molecular reasons underlying their predicted affinities. This section is organized by peptide for clarity. Figure 4 provides a detailed analysis of the binding modes between the peptides and antibodies.
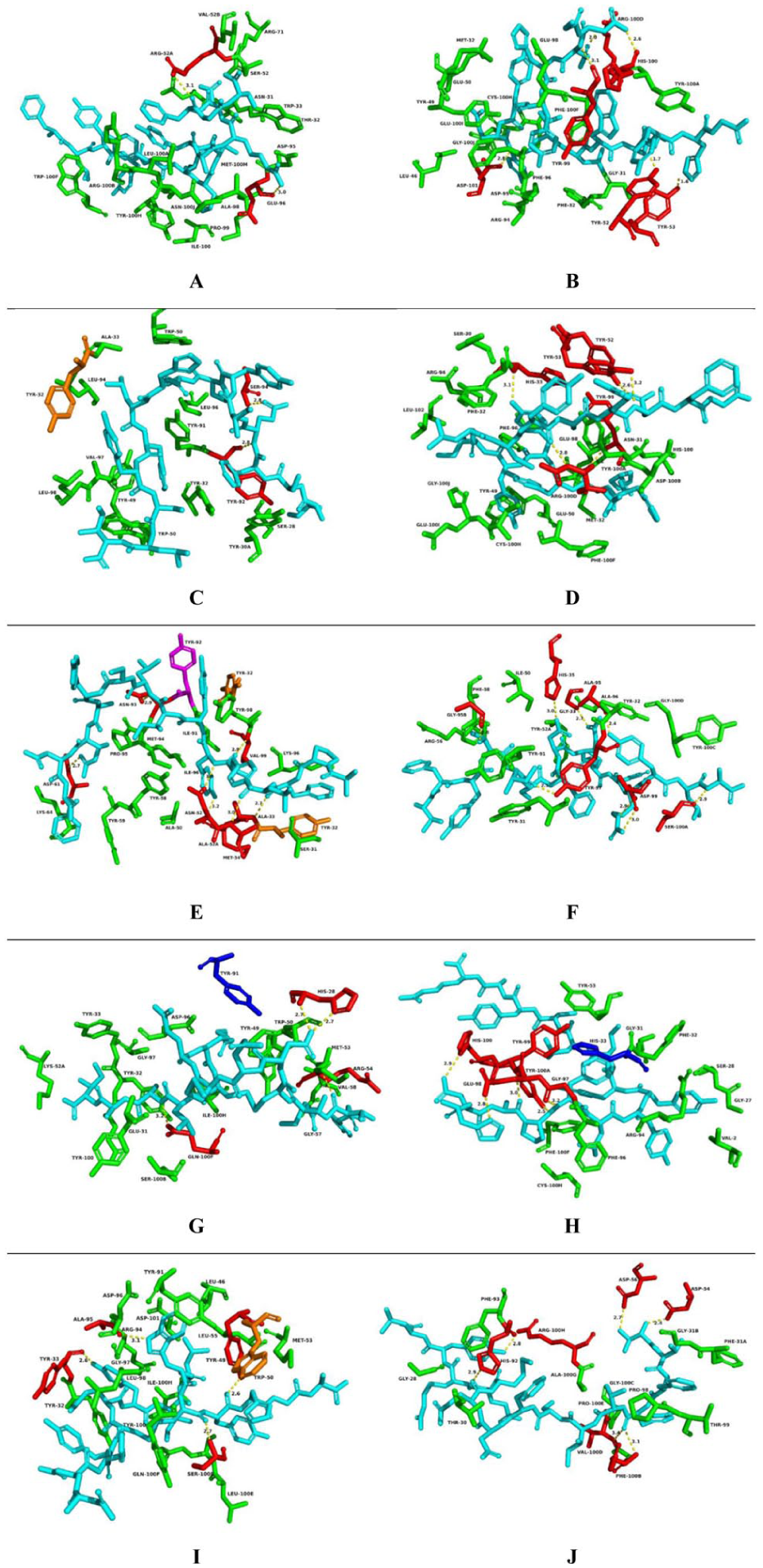
Details of intermolecular interactions for Ag-Ab complexes. (A) Docking result of the DH511.12P Fab (PDB: 5u3n), interacting with Pep I. (B) Docking result of anti-HIV-1V3 Fab 3074 (PDB: 3mly), interacting with Pep II. (C) Docking result of human protective antibody CIS43 (PDB: 6b5m), interacting with Pep III. (D) Docking result of anti-HIV-1 V3 Fab 3074 (PDB: 3mly) interacting with Pep IV. (E) Docking result of Gantenerumab fab fragment (PDB: 5csz) interacting with Pep V. (F) Docking result of Fab311 complex (PDB: 6axk) interacting with Pep VI. (G) Docking result of the antibody 7B2 (PDB: 4ydv) with Pep VII. (H) Docking result of anti-HIV-1 V3 Fab 3074 (PDB: 3mly), interacting with Pep VIII. (I) Docking result of the antibody 7B2 (PDB: 4ydv) interacting with Pep IX. (J) Docking result of the broadly neutralizing anti-HIV-1 antibody 2F5 (PDB: 1tji), interacting with Pep X.
Pep I interact only with the heavy (H) chain of the antibody (PDB: 5u3n), establishing hydrophobic contacts with 16 different amino acid residues. In addition, it forms 2 hydrogen bonds with residues ARG52 and GLU96, at donor-acceptor distances of 3.1 Å and 3.0 Å, respectively. The extensive network of hydrophobic contacts between this peptide and the antibody seems to explain its position as the fourth best prediction among the analyzed peptides.
Pep II, IV, and VIII demonstrated better predicted affinity (lower I_sc values) with the same antibody (PDB: 3mly). They interact with both the light (L) and heavy (H) chains of the antibody, showing a predominance of interactions with the residues of the heavy chain. Pep VIII presented the highest predicted affinity (lowest score: −637.308), followed by Pep IV (−631.192) and Pep II (−573.489). These 3 peptides interact similarly with the target (antibody) through hydrophobic contacts and hydrogen bonds, except Pep VIII, which also interacts via cation-pi interactions, that is, noncovalent interactions between the face/plane of an aromatic ring and an adjacent cation. Despite Pep VIII interacting with the fewest residues (16 residues), the nature of the interactions established contributes to its higher predicted affinity. Notably, the antibody residues ARG94, CYS100H, GLU98, HIS100, PHE100F, PHE32, PHE96, TYR100A, TYR53, and TYR99 are involved in interactions with all 3 peptides, demonstrating their importance for the binding mode of the antigens.
Pep II establishes 6 hydrogen bonds at distances ranging from 1.6 to 3.1 Å, with residues ARG100D (2.0 Å), ASP101 (2.8 Å), HIS100 (2.6 Å), TYR52 (1.7 Å), TYR53 (1.6 Å), and TYR99 (3.1 Å). Pep IV forms 5 hydrogen bonds at distances ranging from 2.6 to 3.2 Å with the residues ARG100D (2.8 Å), HIS33 (3.1 Å), TYR52 (2.6 Å), TYR53 (3.2 Å), and TYR99 (3.2 Å). Pep VIII forms 5 hydrogen bonds at distances between 2.8 and 3.2 Å with the residues GLU98 (2.8 Å), GLY97 (3.2 Å), HIS100 (2.9 Å), TYR100A (2.5 Å), and TYR99 (3.0 Å). Although Pep II establishes one additional hydrogen bond when compared with Pep IV and VIII, its binding mode with the target results in a lower predicted affinity when compared with the other peptides, placing it at the eighth best position. In contrast, the binding modes of Pep VIII and Pep IV achieved the first and second highest predicted affinities, respectively, within the evaluated peptide series.
Pep III interacts with both the light (L) and heavy (H) chains of the antibody (PDB: 6b5 m), showing a predominance of interactions with the light chain. The primary type of interaction is hydrophobic contact, besides a pi-stacking interaction with TYR32 residue, totaling 14 different amino acid residues. In addition, this peptide forms 2 hydrogen bonds with SER94 and TYR92 residues, both with 2.8 Å distance between donor and acceptor atoms. The binding mode for the Ag-Ab complex achieves a predicted affinity of −579.634, ranking it as the seventh best score in the series.
Analysis of Pep V interactions with the antibody (PDB: 5CSZ) reveals the involvement of both the heavy (H) and light (L) chains. The peptide establishes hydrophobic and polar contacts with a total of 24 distinct amino acid residues. Residues such as ALA33, ALA52, ASN93, TYR32, TYR92, and VAL99 interact with the antigen through several kinds of contacts, including hydrophobic (T-stacking and pi-stacking interactions) and polar contacts (hydrogen bonds). The distances between the donor and acceptor atoms of the hydrogen bonds ranges from 2.7 to 3.2 Å, with residues ALA33 (2.7 Å), ALA52A (3.0 Å), ASN93 (2.9 Å), ASP61 (2.7 Å), MET34 (3.0 Å), and ASN52 (double simultaneous interactions at 3.0 Å and 3.2 Å). Shorter hydrogen bonds are typically related to stronger interactions, thus providing greater stability (lower values of I_sc) for Ag-Ab complexes. Pi-stacking interactions (noncovalent interactions between aromatic rings, usually parallel to each other) with the TYR32 residue of the H chain, and T-stacking (noncovalent interactions between aromatic rings, usually in a perpendicular T-shape) with the TYR92 residue of the H chain, besides interactions with 17 other residues through hydrophobic contacts, contribute to enhance the predicted affinity. These interactions result in the third best (more negative) score among the studied peptides.
Pep VI interacts with both the light (L) and heavy (H) chains of the antibody (PDB: 6axk), with a greater predominance of interactions with the heavy chain. This molecule establishes hydrophobic contacts with 16 different amino acid residues and forms 8 hydrogen bonds at distances ranging from 2.4 to 3 Å. The list of interacting residues consists of ALA95 (2.4 Å), ASP99 (2 simultaneous bonds at distances 2.9 and 3.0 Å), GLY33 (2.7 Å), GLY95B (2.6 Å), HIS35 (3.0 Å), SER100A (2.9 Å), and TYR97 (2.5 Å). The binding mode for the Ag-Ab complex achieves a score of −599.219, representing the sixth best predicted affinity compared with other peptides.
Peptides VII and IX interact with both the light (L) and heavy (H) chains of the same antibody (PDB: 4ydv). The residues ASP96, GLN100F, GLY97, ILE100H, MET53, SER100B, TRP50, TYR100, TYR32, TYR33, TYR49, and TYR91 interact with both peptides through hydrophobic contacts and hydrogen bonds. Pep IX has a better (more negative) predicted affinity when compared with Pep VII, establishing hydrophobic contacts with 16 different amino acid residues, besides a pi-stacking interaction with the TRP50 residue. In addition, Pep IX forms hydrogen bonds at distances ranging from 2.6 to 3.1 Å, with the residues ALA95 (3.1 Å), SER100B (2.7 Å), TYR33 (2.6 Å), and TYR49 (2.6 Å). Pep VII, on the contrary, establishes hydrophobic contacts with 17 different residues, besides a cation-pi interaction with the TYR91 residue, and forms 4 hydrogen bonds at distances ranging from 2.7 to 3.3 Å with the residues ARG54 (3.3 Å), GLN100 F (3.2 Å), and HIS28 (2 simultaneous bonds at 2.7 Å). The binding modes of these peptides with the target yield scores of −572.978 and −529.054 for Pep IX and VII, respectively, placing them as the lowest (10th) and second-lowest (9th) in terms of predicted affinity, when compared with the other peptides.
Pep X establishes contacts with both the light (L) and heavy (H) chains of the antibody (PDB: 1TJI), with interactions predominantly occurring within the H chain. This peptide engages in hydrophobic contact with 14 different amino acid residues, and forms 6 hydrogen bonds, with distances ranging from 2.4 to 3.4 Å, involving the residues ARG100H (2.8 Å), ASP54 (2.4 Å), ASP56 (2.7 Å), HIS92 (2.9 Å), PHE100B (3.1 Å), and VAL100D (3.4 Å). The binding mode for the Ag-Ab complex achieves a score of −600.882, ranking as the fifth best score when compared with the other peptides.
Enzyme-linked immunosorbent assay
Each peptide was evaluated individually and in pooled formats using an ELISA assay against human serum samples (n = 78), comprising 50 LT-positive patients and 28 confirmed LT-negative healthy individuals. Assay conditions were optimized prior to testing. Peptide concentrations ranged from 100 to 600 ng, and serum dilutions were tested from 1:450 to 1:900. The results are presented in Figure 5 and Table 3.
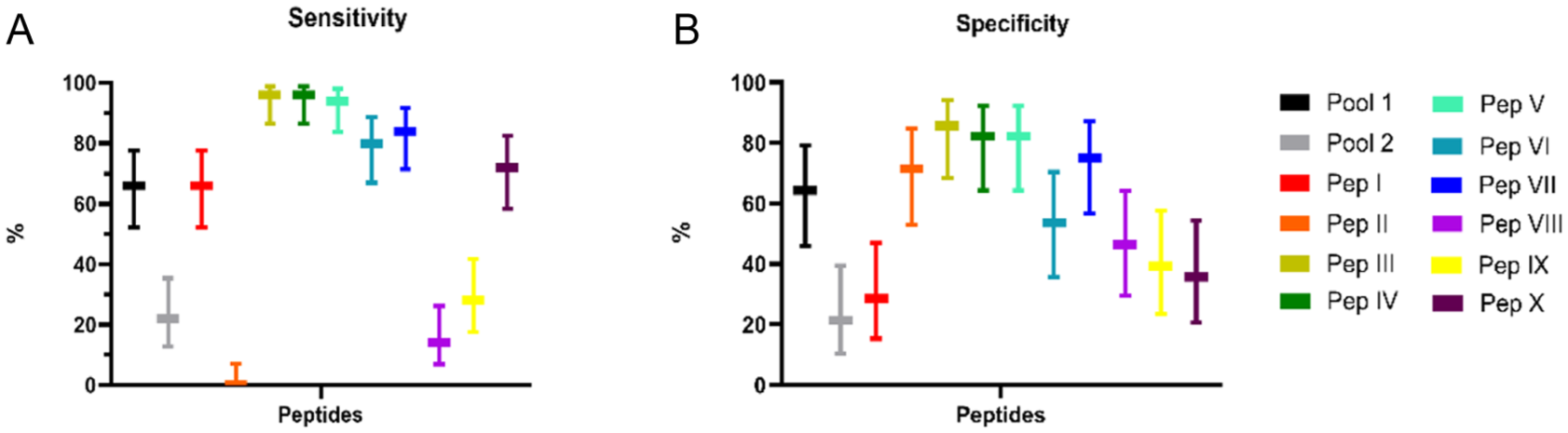
The sensitivity (A) and specificity (B) percentages of different peptide pools and individual peptides when tested against human sera.
Performance of ELISA employing synthetic peptides and sera of patients with cutaneous leishmaniasis and healthy control.

Legend: Se = sensitivity; Sp = specificity; NPV = negative predictive values; PPV = positive predictive values; AUC = area under the curve; CI 95%: Confidence Interval. ROC curves were constructed to determine the ELISA sensitivity (Se), specificity (Sp), and area under the curve (AUC).
The performance metrics of the diagnostic test were calculated using a module (https://www.openepi.com/DiagnosticTest/DiagnosticTest.htm) that compares the test results to a recognized reference standard, known as the gold standard. This approach requires a true diagnostic outcome. The module classified the test results and calculated sensitivity, specificity, PPV, and NPV. When there were multiple result thresholds, a ROC curve was created to show the trade-off between true-positive and false-positive rates. These metrics were used to assess the accuracy and reliability of the diagnostic test.
The table displays the performance of the ELISA test using synthetic peptides for diagnosing CL, evaluated through sensitivity (Se), specificity (Sp), NPVs, PPVs, and the area under the ROC curve (AUC). Pool 1 presented a detection percentage 3 times higher (Se: 66%, Sp: 64.29%, AUC: 0.6500), than Pool 2 (Se: 22%, Sp: 21.43%, AUC: 0.8146). Peptide I exhibited a sensitivity of 66% but demonstrated low specificity (28.57%). Peptide II shows effective excluded noninfected cases (Sp: 71.43%), but did not detect true positives. Peptides III, IV, and V demonstrated strong diagnostic potential, with high sensitivity and specificity (III: Se 96%, Sp 85.71%, AUC 0.9493; IV: Se 96%, Sp 82.14%, AUC 0.9318; V: Se: 94%, Sp 82.14%, AUC 0.951). Peptide VI demonstrated moderate performance (Se: 80%, Sp: 53.57%, AUC: 0.7432), whereas Peptide VII exhibited a better balance between sensitivity and specificity (Se: 84%, Sp: 75%, AUC: 0.8968). Peptides VIII and IX exhibited limited diagnostic performance, characterized by low sensitivity and specificity. In contrast, peptide X demonstrated a sensitivity of 72%; however, its specificity was notably low, yielding only 35.71%.
Comparing in vitro versus in silico
Figure 6 presents the correlation analysis between the in silico predictions (SnugDock docking scores) and the in vitro ELISA results, offering insights into the predictive capacity and limitations of computational modeling in representing biological interactions, particularly for Ag-Ab complexes.

(A) Comparison between Interface Score (I_sc) and Sensitivity Across Various Peptides (B) Comparison between Binding Affinity and Sensitivity of Synthetic Peptides for Ag-Ab complexes.
Figure 6-A illustrates the correlation between the interface score (I_sc) and sensitivity for peptides, evidencing that peptides with higher sensitivity tend to have better (more negative) values of I_sc, that is, indicating a stronger interface interaction for the Ag-Ab complex. For instance, Pep IV and Pep V, both yellow coloreds, exhibit high sensitivity, more negative I_sc, and a high number of interactions. Moreover, Pep III and Pep VII, despite having similar sensitivity, display distinct interaction scores, suggesting a more complex relationship which may involve additional structural and molecular environmental factors.
Figure 6B illustrates how peptide sensitivity relates to predicted binding affinity, using 3 variables: Sensitivity, dG_separated_dSASA × 100, and I_sc. The descriptor dG_separated_dSASA × 100 represents the binding energy per unit interface area, scaled by 100 to fit the units in the score file. A general trend could be observed, where more negative values of dG_separated_dSASA × 100 (binding energy), correlate with more negative (better) I_sc values (indicating higher predicted affinity) and increased sensitivity, as observed with most peptides.
Notably, peptides II, III, VII, and VIII represent exceptions to this trend, as their predicted binding affinities did not consistently correlate with the experimentally observed sensitivities. For example, Pep III stands out as the peptide with the largest difference in binding energy between its Best and Worst I_sc values. This suggests that Pep III’s binding affinity is highly variable, depending on specific conditions, making it a unique case where the usual relationship between binding energy, sensitivity, and predicted affinity seems not to apply. On the other hand, Pep V and Pep IX show a significant difference in binding energy (dG_separated_dSASA × 100) between their Best and Worst I_sc values. Despite this variation, they still follow the general trend where more negative (better) I_sc values, which indicate higher predicted affinity, are associated with more stable (negative) binding energy. For Pep V, this trend is similarly reflected in its increased sensitivity.
Discussion
In recent years, advancements in technology and science have led to the accumulation of extensive data in the fields of immunology, proteomics, and genomics, now widely available through various public databases.25,26 Immunoinformatics has emerged as a vital field to meet the growing demand for computational approaches capable of handling and analyzing these large-scale datasets. It employs a combination of statistical, computational, mathematical, and biological methods to explore immune system functions in depth.27,28 Positioned at the intersection of computer science and experimental immunology, this research field leverages validated immunological evidence data to establish structural and functional signatures. These patterns serve as the foundation for developing predictive tools that provide deeper insights into immune responses in different states, including health, disease and recovery. 28 Furthermore, immunoinformatics has proven its potential in antigen identification for vaccine design and diagnostic applications in parasitic infections. This approach has significantly reduced the time and cost associated with exploration while enhancing diagnostic performance, particularly concerning specificity, sensitivity, and reproducibility.29,30
The aim of this study was to assess leading state-of-the-art methodologies by integrating immunoinformatics with traditional in vitro analysis techniques. This combined approach was used to investigate the interaction dynamics between 10 selected peptides retrieved from the L braziliensis proteome through in silico methods, and different IgG isotypes using both in silico (docking) and in vitro (ELISA) assays with human sera. The selection of peptides analyzed in this study is supported by nearly a decade of ongoing research by our group, as documented in previous publications.7,8 These sequences have undergone validation through both in silico and in vitro methods, including ELISA assays, which revealed distinct immunoreactivity profiles when tested individually and in peptide pools against sera from patients with tegumentary leishmaniasis.
Docking calculations, comprising more than 1 240 000 individual results characterized by 45 structural descriptors, predicted the binding affinities and provided a comprehensive interaction profile. Peptides I, IV, V, and VIII originated from the same hypothetical protein LbrM.34.3630 (a polypeptide found to include a zinc finger and whose T. brucei ortholog localizes to the cytoplasm) exhibited the highest predicted binding affinity (lowest interface scores): Pep VIII (−637.31), Pep IV (−631.19), Pep V (−629.84), and Pep I (−623.69). Among them, Pep IV consistently shows the most favorable and stable interactions, with the lowest variability. Pep VIII and Pep IV, the top 2 score values, respectively, showed high affinity in their interaction with the anti-HIV-1V3 Fab 3074 antibody (PDB: 3mly), suggesting enhanced stability in forming the antigen-antibody (Ag-Ab) complex in these specific cases.
Figure 2 (heatmap) illustrates variations in Ag-Ab interaction strength based on SnugDock descriptors, highlighting key contributors to binding performance. Specifically, dSASA_hphobic, dSASA_int, and dSASA_polar were identified as primary descriptors, reflecting hydrophobic, buried, and polar solvent-accessible interface areas, respectively. These descriptors offer comprehensive insights into the interaction properties between the peptides and antibodies. Figure 3 demonstrates that increased hydrophobic and polar surface areas correlate with lower interface scores, indicative of more stable interactions. A negative correlation between dSASA_int and I_sc suggests that a larger buried interface area enhances complex stability, reinforcing the importance of contact surface in antibody-peptide interactions. Lower I_sc values are achieved by stabilizing the complex, which is then influenced by this. Antigen-antibody complexes exhibit a significant increase in binding affinity due to increasing dSASA_int, which is reflected in decreasing I_sc values.
It is worth mentioning that there is a notable absence of comparable studies in the literature that utilize the same range of structural descriptors for peptide-antibody docking analyses, especially those employing SnugDock. This gap emphasizes the originality of our findings and underscores the significance of this study in enhancing the structural understanding of antigen-antibody interactions at the molecular level.
Regarding the mapping of interactions between peptides and antibodies with the highest score values, it is evident that these peptides interact with both the light and heavy chains of antibodies through hydrophobic and polar contacts. Based on the presented results, it is evident that the score value can be influenced in several ways by the mode of interaction established between the antigen and antibody (Ag-Ab). This interaction mode depends on the structural chemical nature of the antibodies under analysis, specifically the types of residues present at the interaction interface with the antigen (peptide). In this regard, studying protein-peptide interfaces and their constituent residues is crucial for a more detailed understanding of the molecular mechanisms involved in these peptide interactions. Furthermore, identifying these interfaces and residues can provide valuable insights for developing new targets for therapeutic interventions. Therefore, it is essential to conduct a thorough analysis of protein interfaces to enhance the understanding of molecular biology and to advance the development of new therapies with greater specificity and efficacy. 31
Conte et al 32 observed in their published study that buried residues are more apolar compared with other residues present at the partially solvent-accessible interface. Conversely, while the polar surface is composed of neutral and charged groups, the interfaces are deficient in negatively charged groups compared with the rest of the protein surface, although they are not deficient in positively charged groups. Side-chain atoms contribute most of the interface area compared with backbone atoms; however, the contribution of the backbone cannot be overlooked, especially the oxygen atom in the carbonyl group. Non-protein groups are generally irrelevant in most cases, except in some complexes where immunosuppressive drugs or metal ions are present at the interface. Understanding these characteristics can be useful for designing compounds that interact with these interfaces, either as drugs or as therapeutic proteins or peptides.
The analysis of the ELISA results reveals that Pool 1 achieves a detection rate 3 times greater than that of Pool 2. This finding is consistent with previous studies on the immunogenicity of these peptides, as Pool 1 comprises peptides that demonstrated a specific response profile in in vitro assays by stimulating cells from patients with leishmaniasis. In contrast, Pool P2 comprises peptides that showed an unspecific profile, stimulating both cells from the leishmaniasis group and the control group. 8 Therefore, the combination of peptides in Pool P1 demonstrated greater efficacy in differentiating between the sample groups.
Pep III, Pep IV, and Pep V showed excellent performance, with high sensitivity (96%, 96% and 94%, respectively) and high specificity (86%, 82%, and 82%, respectively), in detecting IgG from human sera. In a study conducted by Link et al, 33 3 peptides were identified for the diagnosis of CL, and a sensitivity of 79% was observed using ELISA. Menezes-Souza et al 34 evaluated the performance of 3 linear B-cell epitope peptides and the recombinant heat shock protein 83.1 (rHSP83.1) against serum samples from patients with CL using ELISA. Their results showed sensitivities of 95.55% for rHSP83.1, 71.11% for peptide 01, 64.44% for peptide 02, and 95.55% for peptide 03. Salles et al 35 reported that a linear B-cell epitope (MQKDEESGEFKCEL), derived from the conserved Leishmania SMP-3 protein, exhibited strong diagnostic performance in ELISA assays for human tegumentary leishmaniasis, reaching a sensitivity of 94.5% and a specificity of 92.5%, without cross-reactivity in sera from individuals with unrelated diseases. Medeiros et al 36 examined the diagnostic capabilities of recombinant triparedoxin peroxidase (rTryP), expressed in E coli BL21 (DE3) Arctic cells. Their study evaluated serum samples from 70 patients with cutaneous or mucosal leishmaniasis, compared against 70 samples from healthy individuals and cross-reactive controls. The rTryP ELISA test achieved 88.57% sensitivity and 90% specificity in their analysis. Notably, the sensitivity values observed for peptides III, IV, and V surpass those previously reported in similar studies by other authors.33 -36
The results obtained for peptides IV and V in the ELISA assay align with the in silico prediction approaches, where these peptides demonstrated higher predicted affinity (more negative score values) with the set of antibodies analyzed. By comparing the binding affinities and sensitivities of synthetic peptides obtained from SnugDock solutions, with experimental ELISA data, we can evaluate how significantly in silico models match real-world (experimental) antigen-antibody interactions. Overall, the computational predictions of binding energies show a consistent correlation with increased in vitro sensitivity measurements, thereby confirming the reliability and effectiveness of the in silico methods applied for identifying potential peptide candidates. However, exceptions like those found in peptides II, III, and VIII show that there is still room for improving computational models and highlight the importance of validating these predictions with experimental data.
Conclusion
The findings of this study show that using purified antigens is an effective method for diagnosing infectious and parasitic diseases like leishmaniasis. These stable molecules do not need specialized culture media, which improves the reliability of diagnostic tests. The molecular docking results, along with their descriptors, provided insights into how these peptides bind to antibodies. Peptides VIII, IV, and V exhibited higher predicted affinities compared with others. Notably, the interface score (I_sc) correlates well with solvent-accessible surface area descriptors, such as total (dSASA_int), hydrophobic (dSASA_hphobic), and polar (dSASA_polar) areas. The most promising peptides were Pep III, Pep IV, and Pep V, which showed high sensitivity (96%, 96%, and 94%, respectively) and specificity (86%, 82%, and 82%, respectively). Considering both in silico and in vitro results, peptides IV and V stand out as significant for evaluating Ag-Ab complexes. Their high predicted affinity and ability to detect and differentiate in ELISA assays make them strong candidates for future research and clinical applications, particularly in leishmaniasis diagnosis.
This study also highlights opportunities for developing more sensitive, specific, and accessible diagnostic tools for CL, especially in areas with limited lab resources. The synthetic peptides selected through immunoinformatics, particularly peptides IV and V, performed well in both in silico analyses and ELISA assays, suggesting they could serve as reliable biomarkers. These results support early diagnosis and may lead to the creation of multi-peptide diagnostic panels and the exploration of these antigens as vaccine candidates. In addition, the methods used can be adapted to identify new immunological targets for other forms of leishmaniasis or neglected parasitic diseases, making this approach valuable in health research.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322251375244 – Supplemental material for Evaluation of the Antigenic Potential of Epitopes Derived From Leishmania braziliensis
Supplemental material, sj-docx-1-bbi-10.1177_11779322251375244 for Evaluation of the Antigenic Potential of Epitopes Derived From Leishmania braziliensis by NTC Costa, AMS Pereira, CC Silva, ABX Silva, EO Souza, LFGR Ferreira, MZ Hernandes and VRA Pereira in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
Not applicable
Ethical considerations
This study received evaluation and approval from the Ethics Committee in Human Research at the Aggeu Magalhães Institute (Approval code: 11083812.7.0000.5190).
Consent to participate
All participants consented to be part of the study and signed the Informed Consent Form.
Consent for publication
Not applicable
Author contributions
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by CNPq (Universal—MCTI/CNPq/ FNDCT 479 No 18/2021—Faixa B).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data generated or analyzed during this study are included in this published article and its supplementary information files, further inquiries can be directed to the corresponding author.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.