
Abstract
In this study, we used an immunoinformatics approach to predict antigenic epitopes of Zika virus (ZIKV) proteins to assist in designing a vaccine antigen against ZIKV. We performed the prediction of CD8+ T-lymphocyte and antigenic B-cell epitopes of ZIKV proteins. The binding interactions of T-cell epitopes with major histocompatibility complex class I (MHC-I) proteins were assessed. We selected the antigenic, conserved, nontoxic, and immunogenic epitopes, which indicated significant interactions with the human leucocyte antigen (HLA-A and HLA-B) alleles and worldwide population coverage of 76.35%. The predicted epitopes were joined with the help of linkers and an adjuvant. The vaccine antigen was then analyzed through molecular docking with TLR3 and TLR8, and it was in silico cloned in the pVAX1 vector to be used as a DNA vaccine and designed as a mRNA vaccine.
Introduction
Zika virus (ZIKV) is a positive-sense, single-stranded RNA virus that belongs to the Flaviviridae family and was first isolated in 1947 in Uganda. Proteases produced by the virus and its host can cleave the polyprotein into 7 non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) and 3 structural proteins (C, prM, and E). 1 Following the reported outbreak in Brazil in 2015, ZIKV infection spread rapidly. The disease has been associated with microcephaly and miscarriage during the first 2 trimesters of pregnancy, and less frequently with Guillain-Barré syndrome (GBS).2,3
Currently, there is no vaccine or antiviral drug available against ZIKV. 4 Several candidates are in development simultaneously, including traditional inactivated vaccines, live attenuated vaccines, genetically modified subunit protein vaccines, DNA vaccines, and new mRNA vaccines. Most ZIKV vaccines are still in phase I/II trials, and the vaccine constructs contain prM and E antigens as the main immunogen.5-7 In addition, the NS1 protein, which is secreted during viral replication, is the focus of some vaccine studies. It is highly immunogenic, making it a potential target for the development of a vaccine against ZIKV.5-7 Another interesting finding is that the NS2A protein of ZIKV recruits genomic RNA, the structural protein complex prM/E, and the protease complex NS2B/NS3 to the virion assembly site and regulates the morphogenesis of the virus. 8 These targets are recognized as having an important role in virus replication and entry into the cell. 8 In addition, they allow the identification of possible vaccine epitopes of efficacious subunits.
Immunoinformatics has the potential to reduce the amount of time and resources spent in identifying epitopes compared with traditional peptide screening methods, and its usefulness has been demonstrated in the early stages of vaccine projects, as well as in the pre-screening of potential candidates. Among the advantages of peptide vaccines are as follows: their ability to induce humoral and cellular immune responses; the absence of viral material in the vaccine formulations; the possibility of large-scale production; and that they can be stored in a lyophilized form, thus eliminating the need for a cold chain.9,10 The CD8+ T-cell response lyses infected cells through perforin, and granzymes induces apoptosis and can inhibit infection at various stages in the virus cycle, which are important requirements for vaccine antivirals. 10
Thus, the overall objective of this study was to predict and evaluate the immunogenic activity of computationally predicted peptides derived from ZIKV proteins and to design novel multi-epitope DNA and mRNA vaccines against ZIKV. In this context, CD8+ T-cell and B-cell epitopes were predicted based on the criteria of having conserved, immunogenic, and nontoxic properties, antigenic amino acid sequences, and broad population coverage. A novel DNA and mRNA vaccine candidates were then designed based on this set of epitopes.
Materials and Methods
Obtaining the ZIKV protein sequences
The amino acid sequences of the prM, NS1, and NS2A proteins of the ZIKV isolate from French Polynesia (GenBank accession: KJ776791) were retrieved from the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). In addition, another 21 ZIKV sequences (Supplemental Table S1) were used for comparison purposes, comprising sequences from Africa, Asia, the Pacific, and the Americas.11,12
Prediction of B-cell epitopes
B-cell epitope prediction was performed using the antibody epitope prediction tool (http://tools.iedb.org/bcell) and incorporated in the Immune Epitope Database and Analysis Resource (IEDB). Standard parameters for linear B-cell epitopes were used, based on the Parker hydrophilicity prediction, 13 the Emini surface accessibility prediction, 14 Kolaskar and Tongaonkar antigenicity, 15 and the Karplus and Schulz flexibility prediction 16 methods.
Prediction of T-cell epitopes
The prediction of CD8+ T-cell epitopes from the ZIKV proteins was performed using the MHC-I binding prediction tool, available at IEDB (http://tools.immuneepitope.org/main/index.html). The IEDB recommended method was used, and the epitopes with percentile ⩽1 were used in further analysis.17-19
A set of 27 alleles (HLA-A*01:01, HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*03:01, HLA-A*11:01, HLA-A*23:01, HLA-A*24:02, HLA-A*26:01, HLA-A*30:01, HLA-A*30:02, HLA-A*31:01, HLA-A*32:01, HLA-A*33:01, HLA-A*68:01, HLA-A*68:02, HLA-B*07:02, HLA-B*08:01, HLA-B*15:01, HLA-B*35:01, HLA-B*40:01, HLA-B*44:02, HLA-B*44:03, HLA-B*51:01, HLA-B*53:01, HLA-B*57:01, and HLA-B*58:01) were used from the MHC I group, recommended by the tool, to identify epitopes 8 to 11 amino acids long.
Immunogenicity assessment
The MHC I immunogenicity prediction tool (http://tools.immuneepitope.org/immunogenicity/) was used to assess the ability of MHC I peptide complexes (pMHC-I) to induce T-cell activation. 20 The epitopes predicted in the previous step were used as input, and the default parameters were used. Epitopes with positive scores were used in the conservation analysis.
Epitope conservation analysis
Epitope conservation analysis was performed using the epitope conservancy analysis tool, available at IEDB (http://tools.iedb.org/conservancy/). Epitope conservation assesses the degree of similarity between the epitopes and a set of sequences of interest, 21 so that epitopes whose amino acid sequences are conserved in a variety of different strains can be identified. For this analysis, 21 different ZIKV sequences were used (Supplemental Table S1).
Epitope cluster analysis
The analysis of clustering epitopes was performed using the epitope cluster analysis tool (http://tools.iedb.org/cluster/) to prevent more than 1 epitope from being predicted in the same region and counted as different epitopes. This analysis was based on the identity of the epitope sequences which were grouped in 1 cluster if there was 80% or more shared identity between them. 22 Standard parameters were used in the analysis. The epitope with higher conservation within the cluster was used.
Toxicity analysis
The ToxinPred tool (http://crdd.osdd.net/raghava/toxinpred/) was used to assess whether a predicted epitope was toxic or nontoxic. 23 Standard parameters (prediction method, its threshold value, and e value) were used.
Molecular docking between the epitopes and MHC-I molecules
The tertiary structures of the following MHC-I molecules were obtained from the Protein Data Bank (PDB—http://www.rcsb.org/pdb/home/home.do): 3UTQ (HLA-A*02:01), 3OX8 (HLA-A*02:03), 4NQX (HLA-A*03:01), 4HWZ (HLA-A*68:01), 4I48 (HLA-A*68:02), 6BXP (HLA-B*57:01), and 5TXS (HLA-B*15:01).
Molecular docking was performed to calculate the binding energy between the predicted epitopes and the MHC-I molecules. An incremental protocol optimized for large ligands was used with DINC 2.0 (http://dinc.kavrakilab.org). 24 The predicted complexes were assessed based on their binding affinity, hydrogen bonds, electrostatic interactions, and van der Waals dispersion forces. The three-dimensional (3D) structures of the MHC-I molecules were obtained in the crystallographic form and were then prepared by removing the water around the protein and adding charges to the atoms using Autodock Vina. 25 In addition, the 3D structures of the peptides were determined using the PEP-FOLD 3.5 server (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD/). 26 To visualize the formed complex, the PyMOL tool (https://pymol.org/edu/) was used. Table S1 in the Supplemental Material shows the PDB accession numbers, along with the box grid center coordinates and the dimensions used in DINC, which were calculated in AutoDock Vina.
Analysis of population coverage
Epitopes with affinities less than or equal to 500 nM were used for the population coverage analysis using the population coverage tool, available at IEDB (http://tools.iedb.org/population/), which calculates the proportion of individuals who have a positive response to a group of epitopes with known MHC restriction. 21 Population coverage was assessed for the population of the world, and also for West Asia, Northeast Asia, South Asia, Southeast Asia, Southwest Asia, Europe, East Africa, West Africa, Central Africa, North Africa, South Africa, East India, North America, and South America populations.
Design of the vaccine antigen
For the construction of a subunit vaccine, the epitopes predicted in the previous steps that showed conservation, immunogenicity, nontoxicity, significant population coverage, and strong binding affinity were used. The vaccine antigen was designed as follows: Kozak sequence with the Start codon, 7xHis-tag, 8xGly-tag, EAAAK linker, Adjuvant (pan HLA DR-binding epitope), EAAAK linker, Adjuvant (Beta-defensin), multi-epitope sequence separated by AAY and KK linkers, TAT sequence, and the Stop codon. BamHI, XhoI, KpnI, HindIII, EcoRI, and NotI restriction sites were also added to facilitate cloning (Figure 2). The physicochemical properties of the vaccine were evaluated using the ProtParam tool (https://web.expasy.org/protparam/). 27 Antigenicity was assessed using the VaxiJen 2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). 28 Allergenicity was analyzed using the AllerTOP v.2 server (https://www.ddg-pharmfac.net/AllerTOP/). 29 As the vaccine was designed as a combination of different epitopes, the following servers were used predict the tertiary or secondary structure of the vaccine protein and thereafter validate the structure: RaptorX (http://raptorx.uchicago.edu/), 30 GalaxyRefine (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE), 31 PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/), 32 SWISS-MODEL (https://swissmodel.expasy.org/), 33 and PROCHECK (https://servicesn.mbi.ucla.edu/PROCHECK/). 34
Molecular docking between the multi-epitope vaccine and human immune receptors
Molecular docking was performed to assess the interactions between the multi-epitope vaccine and human immune receptors. Therefore, the HADDOCK 2.4 server (https://wenmr.science.uu.nl/haddock2.4/) 35 was used to assess the docking between the vaccine antigen with TLR3 (PDB ID: 1ZIW) and TLR8 (PDB ID: 3W3G). The lowest HADDOCK score is the most significant score for the docking analysis. To visualize the formed complex, PyMOL (https://pymol.org/edu/) was used. Before molecular docking analysis, active residues (directly involved in the interaction) and passive residues (surrounding residues) for TLR3 and TLR8 were determined using the CPORT server (https://alcazar.science.uu.nl/services/CPORT/). 36
Molecular mechanics and dynamics simulations
The molecular mechanics/general born surface area (MM/GBSA) method was used to evaluate the binding free energy involved in the interactions between the vaccine antigen and the human immune receptors TLR3 and TLR8. The HawkDock server (http://cadd.zju.edu.cn/hawkdock) was used to employ the MM/GBSA method to predict the binding free energy and decompose the free energy contributions to the binding free energy of the vaccine-receptor complex in per-residue. The ff02 force field, the implicit solvent model, and the GBOBC1 model (interior dielectric constant = 1) were used. In the analysis, 5000 steps with a cutoff distance of 12 Å for van der Waals interactions (2000 cycles of steepest descent and 3000 cycles of conjugate gradient minimizations) were used to minimize the system.
The molecular dynamics (MD) simulations were carried out by using the iMODS server (https://imods.iqf.csic.es/), which employs internal coordinates normal mode analysis (NMA). This analysis assesses the structural dynamics of the vaccine-receptor docked complexes, including the molecular motion determination, regarding its deformability, B-factor, eigenvalues, variance, covariance map, and elastic network. All parameters were set to default.
In silico cloning of the vaccine
The Codon Adaptation Tool (http://www.jcat.de/) 37 was used for codon optimization of the vaccine antigen. In addition, to help molecular cloning, restriction sites were inserted at the N-terminal and C-terminal ends of the optimized vaccine antigen. Finally, the optimized nucleotide sequence was cloned into the pVAX1 vector with the SnapGene 4.2 tool (https://www.snapgene.com/) to ensure in vitro expression.
mRNA vaccine design
The mRNA vaccine was designed with a 7-methyl(3-O-methyl) GpppG cap in the 5′ end, followed by 5′ UTR from human hemoglobin subunit beta (HBB) gene (NM_000518.5), Kozak sequence, 7xHis-tag, 8xGly-tag, EAAAK linker, Adjuvant (pan HLA DR-binding epitope), EAAAK linker, Adjuvant (Beta-defensin), multi-epitope sequence separated by AAY and KK linkers, TAT sequence, Stop codon, 3′ UTR from HBB gene (NM_000518.5), and a 120-nucleotide long poly(A) tail.
Then, the secondary structure of the mRNA construct, along with its minimal free energy score, was predicted with the Vienna RNA WebServer, which is based on the ViennaRNA package version 2.6.3 (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi).
Results
Prediction of B-cell epitopes
Kolaskar and Tongaonkar antigenicity for prM, NS1, and NS2A proteins
The formation of antigenic epitopes based on physicochemical characteristics was observed using the method of Kolaskar and Tongaonkar (Supplemental Figure S1). The use of the 74 amino acid ZIKV prM protein sequence resulted in the prediction of an 11 amino acid antigenic peptide (Table 1). Likewise, the NS1 sequence of 352 amino acids resulted in the prediction of 17 antigenic peptides, whose length ranged from 6 to 14 amino acids (Table 1). The 150 amino acid NS2A sequence resulted in 4 antigenic peptides, whose lengths ranged from 7 to 21 (Table 1). In addition, the Kolaskar and Tongaonkar method produces the maximum residual score for each amino acid of the prM, NS1, and NS2A proteins. For the 71 amino acid prM protein, 49 had a residual score higher than 1.0. Furthermore, leucine at position 63 found in the antigenic peptide (VIYLVMI60-66) had a maximum residual score of 1.187.
Prediction of CD8+ T-cell epitopes of the ZIKV prM, NS1, and NS2A proteins.
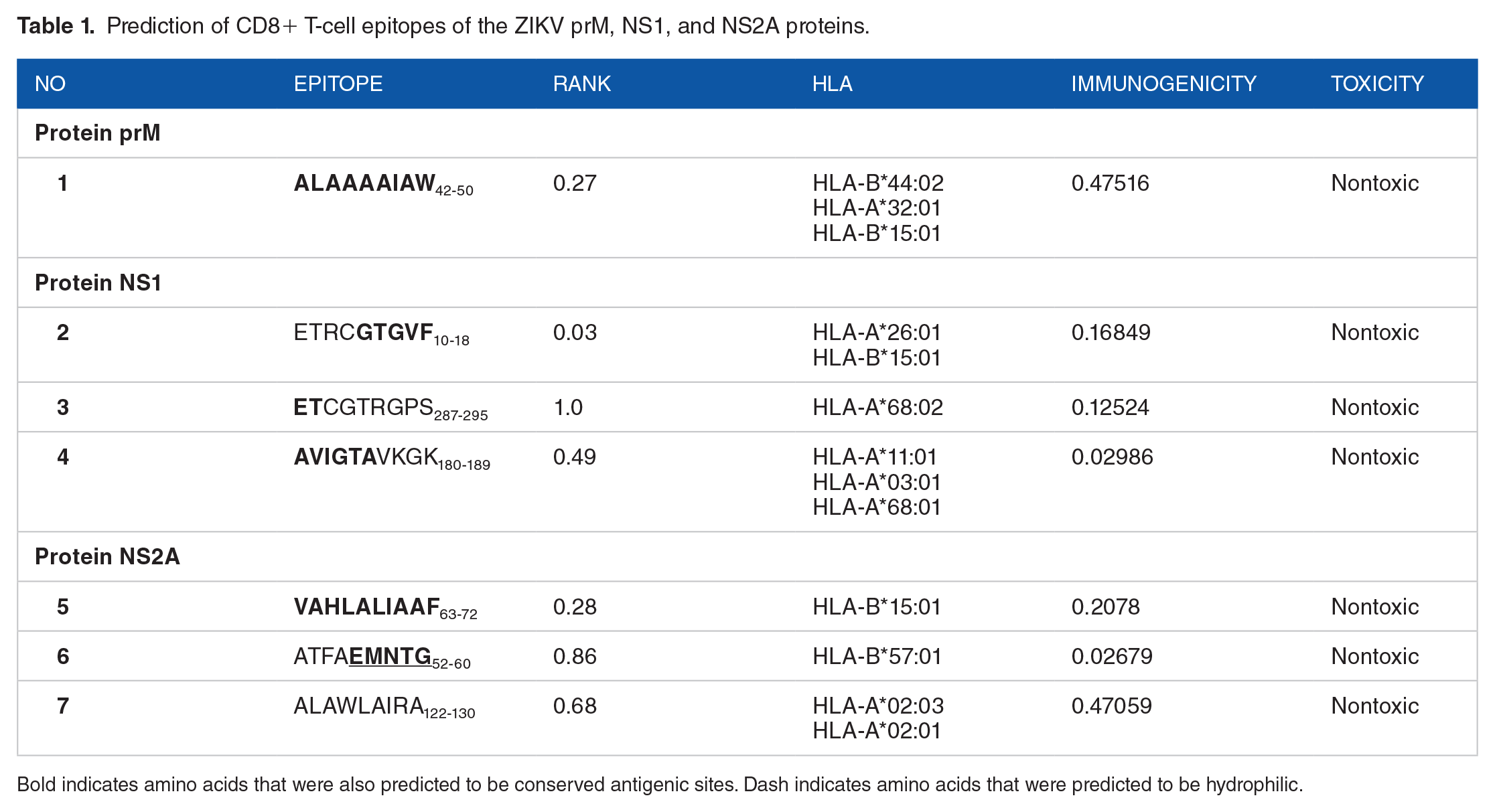
Bold indicates amino acids that were also predicted to be conserved antigenic sites. Dash indicates amino acids that were predicted to be hydrophilic.
Likewise, 219 of the 352 amino acids of the ZIKV NS1 protein were observed to have a residual score >1.0. Threonine at position 85 in the antigenic peptide (VQLTVVV82-88) was found to have a maximum residual score of 1.244. On the contrary, for the ZIKV NS2A protein of the 115 amino acids that were predicted to have a residual score >1.0, isoleucine in position 5, and leucine in position 6 of the antigenic peptides VLVILLM2-8 and LVILLMV3-9, respectively, had the maximum residual score of 1.213. The representation of the minimum and maximum scores can be seen in Tables S2, S3, and S4 in the Supplemental Material. Figure S1 in the Supplemental Material shows the graphical representation of predicted B-cell epitope antigenicity of the prM, NS1, and NS2A proteins based on their sequence position and antigenic propensity. Table 1 includes some amino acids identified in the B-cell prediction of antigenicity as shown in Tables S2, S3, and S4 in the Supplemental Material.
Surface accessibility for prM, NS1, and NS2A proteins
Figure S2 and Tables S2, S3, and S4 in the Supplemental Material show the predicted peptides for the prM, NS1, and NS2A proteins, depending on the sequence position and the surface probability. The minimum surface probability score was 0.05, whereas the maximum surface probability score was 4.278 for the epitope REYTKH22-27 in the prM protein, where Y24 (tyrosine) is a water-accessible surface residue.
In addition, the calculated maximum and minimum surface probability scores were 6.449 and 0.093 for NS1 and NS2A from amino acid position 334 to 339 and 2 to 7 with the hexapeptide sequences RPRKEP and VLVILL, respectively, where Arginine 336 has an increased probability of being on the surface as shown in Figure S2 and Tables S3 and S4 in the Supplemental Material.
Surface flexibility for prM, NS1, and NS2A proteins
Figures S3 and Tables S2, S3, and S4 in the Supplemental Material describe the surface flexibility results for the prM, NS1, and NS2A proteins. A minimum flexibility score of 0.876 was predicted for the heptapeptide sequence YLVMILL62-68, showing it to be a more ordered structure, whereas the maximum score was 1.131 (GSSTSQK53-59) for prM protein.
Instead, the minimum flexibility prediction scores for NS1 and NS2A were 0.908 and 0.879 in the heptapeptide sequences GCWYGME326-332 and ALAWLAI122-128, respectively. It is possible to observe some amino acids identified in the B-cell epitope prediction based on flexibility in Table 1 and also in Tables S2, S3, and S4 in the Supplemental Material.
Parker hydrophilicity prediction for prM, NS1, and NS2A proteins
Figure S4 and Tables S2, S3, and S4 in the Supplemental Material show the predicted epitopes for the prM, NS1, and NS2A proteins based on position sequence and hydrophilicity. The minimum hydrophilicity score was −7.357 for the prM protein, whose heptapeptide sequence was LVMILLI63-69. The maximum hydrophilicity score was 6.014 for the prM protein, whose sequence is GSSTSQK53-59.
In addition, the minimum hydrophilicity scores calculated according to Parker surface hydrophilicity were −2.871 and −6.743, being LIIPKSL239-245 for the NS1 protein, as well as VLVILLM2-8 and LVILLMV3-9 for the NS2A protein. The maximum calculated hydrophilicity score was 7.143 and 5.314 for the heptapeptide sequences ESEKNDT201-207 of the NS1 protein and EMNTGGD56-62 of the NS2A protein, respectively. Table 1 also includes details of some amino acids identified in the B-cell prediction based on hydrophilicity, as shown in Tables S2, S3, and S4 in the Supplemental Material.
Prediction of T-cell epitopes
A total of 898 T-cell epitopes were predicted: prM = 136, NS1 = 397, and NS2A = 365.
Prediction of immunogenicity
The 293 epitopes identified with positive immunogenicity scores were assessed for conservation. Table 1 presents the sequences of the epitopes, along with the immunogenicity score, the rank percentile, and the conservation score.
Toxicity assessment
To eliminate the epitopes with potential cross-reaction against human cells, a toxicity analysis was carried out. After the analysis, none epitopes were considered toxic, that is, without the potential to induce cross-reaction (Table 1).
Molecular docking of ALAWLAIRA122-130 epitope with HLA-A*02:03 peptides
The T-cell epitope exhibited strong binding affinity of hydrogen bonding energy, as shown in Table 2. Likewise, hydrogen bonds were visualized for the ALAWLAIRA122-130 complexes with 3 bonds ranging from 2.7 to 3.3 Å and shared residues (THR 10/H) of the peptide. The complexes showed stability by the formation of hydrogen bonds (Figure 1A).
Epitope interaction with HLA alleles.

The distances between the atoms involved in hydrogen bonds were calculated with the PyMOL program, and the data are angstrom (1 Å = 10−10 m).

Result of molecular analysis of epitopes coupled to HLA-A*02:03 (A), HLA-B*150 (B, C, and D), HLA-A*02:01 (E), HLA-B*57:01 (F), HLA-A*03:01 (G), HLA-A*68:01 (H), and HLA-A*68:02 (I). The molecular surface that forms the groove between the α1 and α2 domains (in gray) is shown. The epitope is represented by lines (magenta), and the red dashed line represents the hydrogen bonds.
Molecular docking of ALAAAAIAW42-50, ETRCGTGVF10-18, and VAHLALIAAF63-72 epitopes with HLA-B*15:01
The HLA complexes docked with ALAAAAIAW42-50, ETRCGTGVF10-18, and VAHLALIAAF63-72 peptides can be seen in Figure 1B, 1C, and 1D and the hydrogen bonds in Table 2. Five hydrogen bonds were formed between the ALAAAAIAW42-50 peptide and the MHC-I protein, with interatomic distances ranging from 2.4 to 3.4 Å. Instead, the second peptide (ETRCGTGVF10-18) formed 8 hydrogen bonds with interatomic distances ranging from 2.4 to 4.1 Å, with common peptide residues such as ARG 3/H. Finally, 7 hydrogen bonds were observed with the VAHLALIAAF63-72 peptide, whose distances ranged from 2.5 to 4.0 Å, observing the sharing of residues from HLA-B*15:01 (TRP 147/HE1).
Molecular docking of ALAWLAIRA122-130 epitope with HLA-A*02:01, ATFAEMNTG52-60 epitope with HLA-B*57:01, and AVIGTAVKGK180-189 epitope with HLA-A*03:01
The molecular docking of the complexes is shown in Figure 1E to G. The complexes showed stability according to the formation of hydrogen bonds (Table 2). Common residues between ALAWLAIRA122-130 (Figure 1E) and THR 73/OG1 of the MHC HLA-A*03:01 of the epitope AVIGTAVKGK180-189 (Figure 1G) were involved in the hydrogen bonds formation. It was also possible to observe 5 hydrogen bonds with the peptide ATFAEMNTG52-60, whose bonds ranged from 3.3 to 3.7 Å (Figure 1F).
Molecular docking of AVIGTAVKGK180-189 epitope with HLA-A*68:01 and ETCGTRGPS287-295 epitope with HLA-A*68:02
Only 1 predicted epitope docked to HLA-A*68:01 (AVIGTAVKGK180-189). Four hydrogen bonds were formed ranging from 3.0 to 4.5 Å (Figure 1H). On the contrary, only 1 predicted epitope docked to HLA-A*68:02 (ETCGTRGPS287-295) (Figure 1I). In this case, there were 6 hydrogen bonds ranging from 2.9 to 4.4 Å. Common interatomic interactions were also observed (THR 5/H-ASN 63/OD1) of the epitopes AVIGTAVKGK180-189 and ETCGTRGPS287-295 with the HLA.
Population coverage
The population coverage of ZIKV epitopes was calculated for the world population and the populations from various regions. Peptide-MHC interactions with IC50 ⩽500 nM were considered. The observed population coverage of the epitopes was above 50% in almost all evaluated regional populations, and in Asia, Europe, India, and America. Central America was the only population with unavailable data on population coverage. Population coverage in Africa and its regions remained around 40% and did not exceed 60%. Nevertheless, population coverage was high in the world population (76.35%; Table 3).
Population coverage of ZIKV predicted epitopes.

The projection of population coverage; baverage number of occurrences of HLA epitopes/combinations recognized by the population; cminimum number of epitope/HLA combinations recognized by 90% of the population.
Design of the vaccine antigen
The design of the final vaccine antigen was based on a sequence of a total of 5 T-cell epitopes and 3 B-cell epitopes that fulfilled all the previously adopted criteria, and combined with linkers. Thus, the β-defensin adjuvant was added to the N-terminal region, with the EAAAK linker, to increase the immunogenicity of the vaccine antigen, which is composed of a long chain of 45 amino acids (GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK). To allow intracellular delivery of the designed vaccine, a TAT sequence (11 aa) was appended to the C-terminus. After the addition of linkers and the adjuvant, the final vaccine antigen had 205 amino acids, which was converted into a DNA vaccine with 644 nucleotides (Figure 2).

Schematic drawing of vaccine construction after addition of linkers and adjuvant.
Antigenicity and allergenicity assessment
Antigenicity prediction using the VaxiJen 2.0 web server was performed including the adjuvant and showed a total protective antigen prediction probability of 0.5924 with the virus model, being characterized as a probable antigen. Using the AllerTOP v.2 server, it was possible to observe that the vaccine would have a non-allergenic nature.
Analysis of solubility and physicochemical properties
The antigen was analyzed for solubility and its physicochemical properties using the ExPASY ProtParam server. The results showed an antigen molecular weight of 15 kDa. The theoretical isoelectric point (pI) value of the antigen was 9.58. Therefore, the antigen is considered to be highly basic. The half-life of the antigen was 30 h in mammalian reticulocytes in vitro, >20 h in yeast, and >10 h in Escherichia coli in vivo. The instability index was 22.80, indicating that it is a stable model. The aliphatic index of the protein was 68.64, confirming its thermostability. 38 Grand average of hydropathy (GRAVY), the large hydropathy mean for the antigen was −0.303, indicating its hydrophilic nature.
Analysis of the structure of the vaccine antigen
The PSIPRED server generated the antigen secondary structure, and among the 205 amino acids, the antigen is made up of 52.7% alpha-helix, 10.2% beta-sheets, and 37.1% loops (Supplemental Figure S5). Furthermore, based on the accessibility of amino acid residues, 53% were predicted to be exposed, 18% to be exposed to the medium, and 28% were predicted to be buried. A total of 5% of residues were found in the scrambled domains.
To determine the tertiary structure of the vaccine, the RaptorX server 39 was used. The structure was then refined (Figure 3) using the GalaxyRefine server. 40 The quality assessment of the vaccine structure model showed that 96.06% of the amino acid residues were in the most favored regions of the Ramachandran plot, which reflects the stereochemical quality of the antigen structure (Supplemental Figure S6).

Tertiary structure of the vaccine antigen. Alpha helices are shown in red, β-sheets in light blue, and loops in gray and green.
Molecular docking between the vaccine antigen with TLR3 and TLR8
A proper association between immune receptor molecules and the antigen molecule is essential to activate immune responsiveness. The HADDOCK 2.4 server was, therefore, used to perform the coupling of the vaccine antigen with the human immune receptors TLR3 and TLR8. These receptors can efficiently induce the immune response after virus recognition. Molecular docking analysis showed interactions between the vaccine antigen and TLR3/TLR8. The TLR3 is shown in magenta, whereas the vaccine antigen is shown in green in Figure 4A and B. The vaccine antigen interacted in the range of 1.6 to 3.7 Å with TLR3 (Figure 4B). Table 4 gives details of the quantitative information of the best fit with the HADDOCK score of −137.3 ± 17.0 (au), which suggests a good binding affinity between TLR3 and the vaccine antigen. The model used suggests a protein surface less exposed to water and close binding proximity, which is indicated by the buried surface area (BSA) of 2736.6 ± 233.6 (Å2).

Construction of vaccine antigen with TLR3 and TLR8. (A and C) Molecular docking of vaccine antigen with TLR3 and TLR8 in 2 cartoon representations. The TLR3 is displayed in magenta, and the vaccine antigen construct is displayed in green; TLR8 is displayed in gray, and the vaccine antigen construct is displayed in green. (B and D) Enhanced image of the interactions of the vaccine antigen complex and the TLR3 and TLR8. The yellow hatched area represents the interactions of the vaccine antigen complex and the TLR3 and TLR8.
Statistics of the docking analysis between TLR3 and TLR8 and the vaccine antigen.

The TLR8 is shown in gray, whereas the vaccine antigen is shown in green in Figure 4C and D. The vaccine antigen interacted in the range of 2 to 3 Å with TLR8 (Figure 4D). Table 4 details the quantitative information of the best fit with HADDOCK score of −75.5 ± 6.4 (au), which suggests a good binding affinity between TLR8 and the vaccine antigen. The model used suggests a protein surface less exposed to water and close binding proximity, which is indicated by the BSA of 1529.5 ± 103.3 Å2.
MM and MD simulations
To assess the binding free energy involved in the interactions between the vaccine and the immune receptors TLR3 and TLR8, the MM-GBSA method was used. The analysis revealed the binding free energy of the vaccine-TLR3 complex as −23.97 kcal/mol. For the vaccine-TLR8 complex, the binding free energy was −39.83 kcal/mol.
To evaluate the stability and physical movements of the vaccine-receptors complexes, MD simulations using the iMOD server were performed. The deformability gives the mobility profiles of the vaccine-receptors complex, in which the highest peaks are associated with the regions of high deformability. It is possible to observe that the deformability of the 2 vaccine-receptor complexes remains stable throughout the simulation with mean values below 0.3 (Figures 5A and 6A).

Molecular dynamics simulation analysis of the vaccine-TLR3 docked complex: (A) the main-chain deformability graph; (B) eigenvalue plot; (C) normal mode variance plot; (D) the covariance matrix; and (E) the elastic network model.
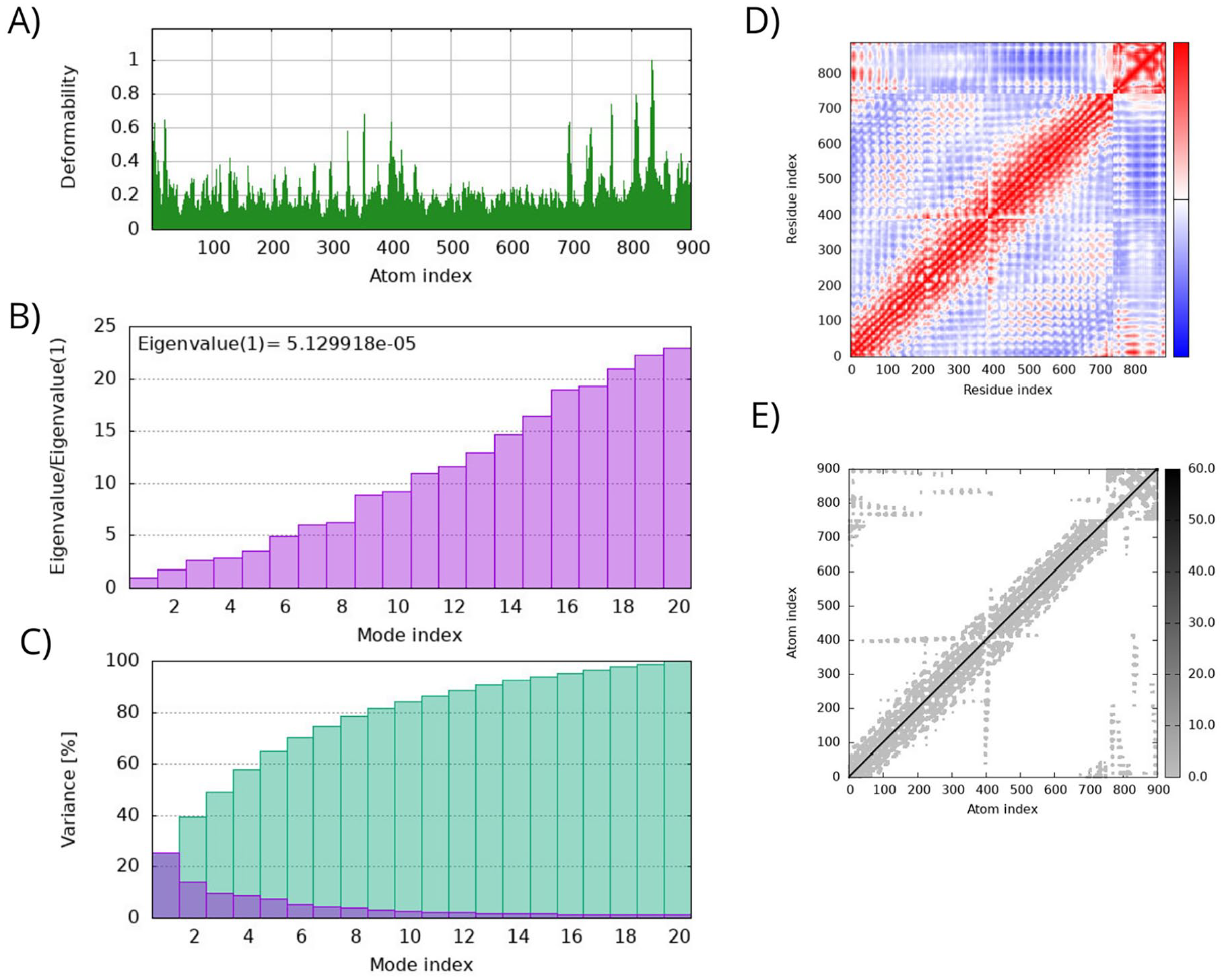
Molecular dynamics simulation analysis of the vaccine-TLR8 docked complex: (A) the main-chain deformability graph; (B) eigenvalue plot; (C) normal mode variance plot; (D) the covariance matrix; and (E) the elastic network model.
The eigenvalue and the variance are inversely linked to each normal mode. The eigenvalue and variance graphs of the vaccine-receptor complexes are represented in Figures 5B, C, 6B and C. The eigenvalue and variance graphs evidence relatively high stiffness of both complexes. The covariance matrix of the 2 complexes shows a great correlation between residues in a complex, which is represented in the red diagonal (Figures 5D and 6D). The elastic maps of the vaccine-receptor complexes also show a great association among the atoms, which represents stable complexes (Figures 5E and 6E).
In silico cloning
To fuse the final vaccine antigen sequence to an expression vector, the codon conversion of the vaccine antigen protein was performed with the Java Codon Adaptation tool. The vaccine antigen sequence was cloned into the pVAX1 expression vector (Figure 7), and then, the structure was generated using the SnapGene program, containing 3575 bp. The pVAX1 plasmid vector has been widely used as a vaccine vector for containing a strong cytomegalovirus promoter for high-level expression of recombinant proteins in mammalian cells, and a bovine growth hormone poly(A) signal for efficient transcription termination and polyadenylation of mRNA. In addition, pVAX1 is consistent with the recommendation of the Food and Drug Administration on the plasmid DNA vaccines for preventive infectious diseases (Docket no. 96N-0400).

In silico cloning of the vaccine antigen into the pVAX1 vector. The light pink area represents the vaccine antigen, and the other colored areas represent the ligands and adjuvant.
mRNA vaccine design
The predicted minimum free energy (MFE) of the secondary structure of the mRNA vaccine construct was −349.30 kcal/mol, and the centroid secondary structure of the mRNA presented a MFE of −328.60 kcal/mol, which suggests that the mRNA vaccine is predicted to be stable (Figure 8A and B). The mountain plots of positional entropy of the MFE structure and the centroid secondary structure support the structural stability of the mRNA construct. This plot is a line graph representation of the MFE structure, the thermodynamic ensemble of RNA structures, and the centroid structure, which shows that the structures overlap to a greater extent, and the mRNA structures show considerable similarities in the loop and stem organization as compared to the thermodynamic ensemble of RNA structures (Figure 8C).

Secondary structure prediction and stability analysis of the mRNA vaccine construct: (A) minimum free energy (MFE) structure of the mRNA vaccine construct; (B) centroid free energy (CFE) of the mRNA vaccine construct. The color scale is based on positional entropy; and (C) a mountain plot representation of the secondary structure of the mRNA vaccine constructs and the positional entropy. Red, green, and blue line graphs represent the MFE structure, the thermodynamic ensemble of RNA structures, and the centroid structure, respectively. The height represents the number of base pairs enclosing the base at 1 position. Peaks correspond to hairpin loops, plateaus correspond to loops, and slops correspond to helices.
Discussion
This study aimed to identify potential epitopes of CD8+ T cells and B cells and to design a novel multi-epitope vaccine candidate using immunoinformatics methods. Novel vaccine identification using computational approaches can be performed with the pathogen genomics/proteomics analysis. 41 In this context, problems with culturing pathogens and with the in vitro expression of antigens can also be avoided. 41 In addition, in silico vaccine candidates have been shown to produce promising in vitro and in vivo results. 42
In this study, we used the structural protein (prM) and 2 non-structural proteins (NS1 and NS2A) from the H/PF/2013 strain of ZIKV from French Polynesia. The NS1 protein, which is secreted during viral replication, is used in vaccine studies and is highly immunogenic, making it a potential target for the development of a vaccine against ZIKV. 5 - 7 The ZIKV NS2A protein is responsible for recruiting genomic RNA, and prM/E and NS2B/NS3 complexes to the virion assembly site and arranging viral morphogenesis. 8 These targets are recognized as having an important role in virus replication and entry into the cell. 8 In addition, they are targets in genomic and immunologic data for the development of vaccines that allow the identification of possible vaccine epitopes of effective subunits.
Data from the literature data show that vaccine projects that include non-structural proteins in addition to structural proteins should be considered as they could induce cytotoxic T cells and polyfunctional helper T-cell responses. 43 In addition, non-structural proteins induce cross-reactions, which are likely to be protective against non-dengue viruses. 43 Therefore, it is important to include non-structural proteins to induce CD8+ T-cell protection in new ZIKV vaccines. 43
Immunoinformatics has been used to predict potential antigenic epitopes of ZIKV proteins for the development of peptide vaccines. Most ZIKV vaccines are still in phase I/II trials, and the constructs contain, as the main immunogen, prM and E antigens, which target neutralizing antibodies.6,7,44 To the best of our knowledge, no vaccine candidate is in phase III, nor licensed for use in flavivirus infection.6,7,44
To establish a bridge between the activation of the immune system and its adaptive immunity cells in ZIKV infection, this study used CD8+ T cells and B-cell epitopes. Among the main immunologic effectors mediated by a vaccine are the antibodies (B lymphocytes), CD8+, and CD4+ T cells, depending on the organism involved.10,45 CD8+ T cells and B cells participate directly in the antiviral response to ZIKV, indicating that cytotoxic CD8+ T-cell responses probably play a role in protecting ZIKV infection, as well as other flaviviruses. 46 On the contrary, the response to neutralizing antibodies can inhibit infection at stages in the virus life cycle, including blocking virus binding to host cells and blocking infection at the fixation stage, thus interfering with pH. In contrast, under certain conditions, some flavivirus-reactive antibodies enhance the infection by non-neutralizing antibodies that bind to the virion and promote the infection and internalization via increased fixation. 10
Therefore, in this study, we performed the prediction of B cells in which we identified epitopes with accessibility, flexibility, hydrophilicity, and antigenic profiles for the prM, NS1, and NS2A proteins of ZIKV. In the predictions of accessibility, flexibility, antigenicity, and hydrophilicity of B cells with the prM protein, we did not identify epitopes with all established criteria. On the contrary, in the predictions of flexibility and hydrophilicity for the NS1 protein, the heptapeptide (ESEKNDT201-207) presented the best results, and the lysine at position 204 was shown to be flexible and hydrophilic. In the analysis with the NS2A protein, the heptapeptide (EMNTGGD56-62) showed good antigenicity and hydrophilicity, especially with the threonine at position 59. Finally, 3 B-cell epitopes were the most promising candidates due to their presence of T-cell epitope residues: ALAAAAIAWLL42-52 (prM), GTGVFVYND14-22 (NS1), and VAHLALIAAFKVRPALLVSFI63-83 (NS2A).
Among the epitopes that can bind MHC I in T-cell prediction for the prM protein of ZIKV, we observed that ALAAAAIAW42-50 was the only one that met all the criteria used in this study. We also identified the epitopes for the ZIKV NS1 protein: ETRCGTGVF10-18, ETCGTRGPS287-295, and AVIGTAVKGK180-189. Finally, for the ZIKV NS2A protein, those that met the requirements for immunogenicity, toxicity, and conservation rate were VAHLALIAAF63-72, ATFAEMNTG52-60, ALAWLAIRA122-130, and MNTGGDVAH57-65.
An evaluation of the prediction of T-cell epitopes showed that they have positive immunogenicity values, suggesting that the peptide-MHC I complexes can be recognized by T cells. These data confirm the immunogenic potential of the epitopes identified.22,47,48 These epitopes have also been identified as nontoxic to humans. In the conservation analysis, we used 21 sequences from Africa, Asia, the Pacific, and the Americas11,12 and found that the epitopes were conserved.
Overall, in silico approaches aid in reducing the number of in vitro trials and providing an important tool in the design of vaccines.49,50 In this study, we observed good interactions and hydrogen bonding using molecular docking analysis. However, further studies are still needed to confirm whether the target (epitope) can interact with the specific region of the HLA macromolecule, observing the affinity and stability of the complex formed. 24
Hydrogen bonds and interactions between the peptide or MHC residues were also observed. The peptide ALAAAAIAW42-50 interacted with the largest number of MHC I alleles of the ZIKV prM protein with high binding affinity to HLA-B*15:01, with 5 hydrogen bonds. In addition to this peptide, it was observed that 2 peptides (ETRCGTGVF10-18 and VAHLALIAAF63-72) had high binding affinities to HLA-B*15:01, with 8 and 7 hydrogen bonds, respectively; that is, we observed that the HLA-B*15:01 allele showed the highest number of hydrogen bonds formed when interacting with these 3 epitopes.
Other interactions were observed in this study with binding affinity to other alleles, namely HLA-A*02:03, HLA-A*02:01, HLA-B*57:01, HLA-A*03:01, HLA-A*68:01, and HLA-A*68:02. Hydrogen bonds play a significant role in the stabilization of the ligand in its receptor, becoming the main parameter in the observations of receptor-ligand interactions. 51 Furthermore, several authors have reported a close association between stability and immunogenicity, in which a more stable peptide-HLA complex correlates with greater immunogenicity.52-54
In addition, we calculated the population coverage of the epitope-binding alleles. Except for Central America, the population coverage was high in all the other populations, including the whole world population, which was above 70%. A T cell will recognize a particular epitope only if the host’s MHC molecule is capable of binding to it due to the MHC restriction. 55 Therefore, this is relevant to predict how the population will react to the vaccine.
A set of 6 epitopes were identified (ALAAAAIAW42-50, ETCGTRGPS287-295, AVIGTAVKGK180-189, VAHLALIAAF63-72, ATFAEMNTG52-60, and ALAWLAIRA122-130) with the affinity of 1 to 3 molecules of HLA-A and/or HLA-B and were immunogenic and conserved in the context of various HLA molecules. Dawson et al 56 concluded that to achieve 80% coverage, 3 to 5 HLA molecules are needed in certain ethnicities. In this respect, the set of epitopes predicted in this study presents high population coverage in the world population (greater than 70%) and also in more specific populations.
To improve the immunogenicity of the vaccine antigen, we inserted adjuvant and linker sequences between the previously predicted epitopes to increase antigenicity. The fact that it has no allergenic properties further confirms its potential as a vaccine candidate.
For the transport of the antigenic protein within the body to be effective, it must interact with immune receptors. Therefore, the vaccine antigen-TLR complex was assessed by molecular docking simulations and was found to be capable of inducing a protective immune response in silico. Toll-like receptors (TLRs) play an important role in immunity, being the first stage of the defense against viral infections. These receptors detect the pathogen-associated molecular patterns (PAMPs), activating innate immunity and regulating the immune response. 57 Previous findings show that TLR stimulation can induce a strong response against viruses that induce type I interferon (IFN) responses. 58 Also, according to the literature, the TLR3 receptor is involved in mediating and signaling responses that provide defense against viral diseases, which can be improved by the use of an adjuvant, 59 so its role is considered vital in generating an antiviral immune response. A study reported that ZIKV activates TLR3 to produce an inflammatory response, which probably dampens the antiviral response. 60
Conclusions
In conclusion, the B-cell epitopes ALAAAAIAWLL42-52 (prM), GTGVFVYND14-22 (NS1), and VAHLALIAAFKVRPALLVSFI63-83 (NS2A) met all the established criteria and also had shared T-cell epitope residues. This study was also able to predict immunogenic, nontoxic, conserved epitopes, with significant population coverage in the world population that presented antigenic sites in prM protein (ALAAAAIAW42-50), NS1 protein (ETRCGTGVF10-18, ETCGTRGPS287-295, and AVIGTAVKGK180-189), and NS2A protein (VAHLALIAAF63-72, ATFAEMNTG52-60, ALAWLAIRA122-130, and MNTGGDVAH57-65), which are capable of binding to MHC I. With these B- and T-cell epitopes, it was possible to design a multi-epitope vaccine candidate that is antigenic, non-allergic, and thermostable, presents good solubility, and can effectively interact with TLRs, which could be tested in further in vitro and in vivo studies.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322241257037 – Supplemental material for Immunoinformatics-Based Design of Multi-epitope DNA and mRNA Vaccines Against Zika Virus
Supplemental material, sj-docx-1-bbi-10.1177_11779322241257037 for Immunoinformatics-Based Design of Multi-epitope DNA and mRNA Vaccines Against Zika Virus by Juciene de Matos Braz and Marcus Vinicius de Aragão Batista in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (grant no. 307128/2022-9) (Bolsas de Produtividade em Pesquisa—PQ), the Fundação de Apoio à Pesquisa e a Inovação Tecnológica do Estado de Sergipe, and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Finance Code 001).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
The individual contribution of each author was performed as follows: Juciene Braz: Conceptualization; Investigation; Methodology; Formal analysis; Visualization; Validation; Writing - original draft. Marcus Batista: Conceptualization; Methodology; Resources; Visualization; Validation; Funding acquisition; Project administration; Supervision; Writing - original draft; Writing- Review and Editing. All authors revised and approved the final manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.