
Abstract
Background:
Human papillomavirus (HPV) causes disease through complex interactions between viral and host proteins, with the PI3K signaling pathway playing a key role. Proteins like AKT, IQGAP1, and MMP16 are involved in HPV-related cancer development. Traditional methods for studying protein-protein interactions (PPIs) are labor-intensive and time-consuming. Computational models are becoming more popular as they are less labor-intensive and often more efficient. This study aimed to develop a deep learning model to predict interactions between HPV and host proteins.
Method:
To achieve this, available HPV and host protein interaction data was retrieved from the protocol of Eckhardt et al and used to train a Recurrent Neural Network algorithm. Training of the model was performed on the SPYDER (scientific python development environment) platform using python libraries; Scikit-learn, Pandas, NumPy, and TensorFlow. The data was split into training, validation, and testing sets in the ratio 7:1:2, respectively. After the training and validation, the model was then used to predict the possible interactions between HPV 31 and 18 E6 and E7, and host oncoproteins AKT, IQGAP1 and MMP16.
Results:
The model showed good performance, with an MCC score of 0.7937 and all other metrics above 88%. The model predicted an interaction between E6 and E7 of both HPV types with AKT, while only HPV31 E7 was shown to interact with IQGAP1 and MMP16 with confidence scores of 0.9638 and 0.5793, respectively.
Conclusion:
The current model strongly predicted HPVs E6 and E7 interactions with PI3K pathway, and the viral proteins may be involved in AKT activation, driving HPV-associated cancers. This model supports the robust prediction of interactomes for experimental validation.
Keywords
Introduction
Persistent infection with high-risk human papillomavirus (HPV) has been implicated in approximately 600 000 cases of cancers of the cervix, oropharynx, anus, vulvovaginal, and penis.1-3 The genome of HPV is divided into 3 parts. The first part is the early region containing the early genes. The early genes E1 and E2 code for the major replication proteins E1 and E2 while E4 and E5 proteins aid in genome amplification. The E6 and E7 proteins are the main oncoproteins that drive carcinogenesis. The second is the late region, containing the late genes L1 and L2, which encode the L1 major and L2 minor capsid proteins. The third part is the long control region (LCR) which is the only noncoding region of the genome, and contains the early viral promoters, enhancers, and the origin of viral DNA replication.4,5
The ability of HPV to cause disease relies on a complex interaction between early viral proteins and host proteins. The carcinogenic transformation of HPV-associated lesions is as a result of deregulation of virus-host cross-talk, leading to over-expression of E6 and E7 viral oncogenes and consequently the accumulation of cellular genetic mutations. 6 The PI3K/Akt signaling pathway has been reported to play a central role in the virus/host cell cross-talk of HPV-positive cancer cells. 7 The AKT, IQGAP1 and MMP16 have all been implicated in HPV-associated carcinogenesis through the PI3K pathway.8-10 Therefore, understanding how HPV proteins and host proteins interact in the disease state will be very useful in elucidating infection mechanisms and unraveling more efficient approaches to managing the disease. This can be done through prevention, risk identification based on genetic profiles, and better drug design. Currently, available experimental approaches for studying protein-protein interactions (PPI) include pull down, 2-hybrid, gel filtration chromatography, isothermal titration calorimetry, Förster resonance energy transfer, luminescent oxygen channeling, reflectometric interference spectroscopy, and other high-throughput biological techniques. 11 Even though these experimental methods have been used to identify PPIs in several model organisms, they are time-consuming and labor-intensive. In addition, their applicability depends on the effectiveness of assay protocols as some may not work on certain classes of proteins. Similarly, experimental methods may miss weak interactions and leave out many transient interactions. 12
Therefore, the use of computational methods to complement experimental methods in predicting PPIs is gaining widespread acceptance as they are less labor-intensive, fast and more efficient. Machine learning (ML), a subfield of artificial intelligence that focuses on using data to learn associations and make predictions, is the most common computational method in use. 13 Generally, ML algorithms have 4 categories: supervised learning, unsupervised learning, semi-supervised learning, and reinforcement learning. 14 Among these methods, supervised learning, which is the focus of this study, is the most used approach. The supervised ML method relevant to PPI predictions is the binary classification approach. This involves the use of binary labeled training data (2 input proteins and their interactions), to construct a model that infers labels for test data (which usually originates from the same distribution as the training data, but without labels). 15 The model then extracts the relevant features from each of the protein representations and uses them for PPI prediction. In supervised ML, training data is used to adjust model parameter settings that tend to predict known interactors accurately. 15 Once trained, the model is expected to predict labels in the training data accurately. The test data, however, serves as unseen data and provides information on how well the model performs beyond the training data. This helps to test the model’s ability to generalize on new data sets. The aim of this study was to adopt a suitable deep learning model to predict HPV and host PPIs.
Materials and Methods
The main data set used for this experiment was from the protocol of Eckhardt et al. They mapped the global network of HPV31 proteins and host PPIs by purifying the complete set of HPV31 proteins in multiple cell lines, followed by mass spectrometry analysis. 16 The data sets had 3 main subsets with interactions between HPV31 and host protein pairs: HEK293 PPIs (405 positives and 393 negatives), C33A PPIs (137 positives and 128 negatives), and Het-1A PPIs (84 positives). The positive interactions were defined in the experiments as those with MiST scores greater than 0.75 on a scale of 0 to 1. The negative interactions were sampled out of the rest as those with MiST scores less than 0.02. The total data set used for the model training, validation and testing was the combined data sets of HEK292 and C33A (1064 PPIs). Out of this, the training set had 744 PPIs, the validation set had 106 PPIs and the test set had 214 PPIs. The Het-1A data set was held as an external validation data set while additional data on HPV18 and host PPIs was obtained from the Viruses.STRING online database, 17 to test the model’s ability to predict other HPV types and host PPIs.
Methods
Obtaining data
The amino acid sequences of the proteins for each interaction in the data set were obtained from UniProt database as FASTA files 18 using a custom python script and a spreadsheet containing their unique UniProt accession numbers. The files for each protein pair were then stored in a working directory.
Preprocessing data
The retrieved data was preprocessed by converting each protein sequence into a suitable data format for the prediction model, using a technique known as tokenization. This was based on the approach used in a similar study, 19 where the protein sequences were broken into 3-gram non-overlapping tokens. The attributes used to describe each PPI data set consist of the labels, Protein_1_bait and Protein_2_prey, which were assigned to the HPV and human host protein pair, respectively, while 0 and 1 were assigned to the pair for interactivity (1 for interaction, 0 for non-interaction). The amino acid sequences were retrieved and modeled as a “3-gram” sequence, where three contiguous amino acids were joined to form a single unit or “word” as done in AI-based natural language processing (NLP). For example, an amino acid “X” preceded by amino acid “Y,” and followed by amino acid “Z,” together form a triplet, “YXZ.” Every triplet in the sequence is then encoded as an integer (1,2,3, . . . [last token]) to produce a series of tokens. To obtain a fixed length of the tokenized protein sequences (1000 elements), the token “0” was reserved for padding the length of the numeric sequences to the left of the vector until the length of 1000 was obtained. If the vector sequence of the protein was longer than 1000, it was truncated, discarding the leftmost elements (the beginning of the amino acid chain) in the sequence (Figure 1). A 3-gram modeling approach performed well in similar instances whereby n-gram modeling in protein informatics was applied using NLP.19,20 Nevertheless, reported n values for n-gram modeling can range from n = 2 up to n = 6.21,22
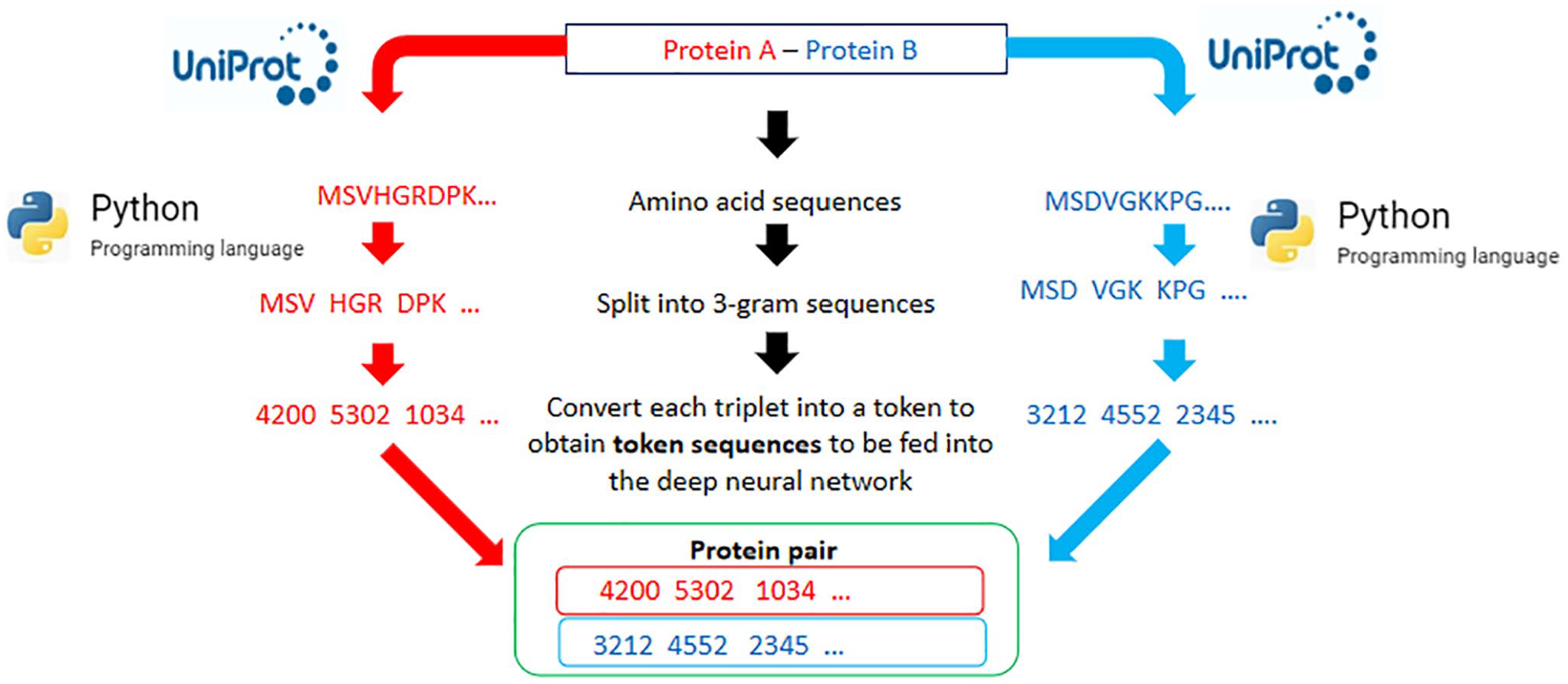
The data preprocessing workflow for training the deep learning model. The protein sequences are obtained from the UniProt online repository and then preprocessed using python programming to obtain tokenized sequences of numerical vectors.
Training
Model training was performed on the SPYDER platform using python libraries: Scikit-learn, Pandas, NumPy, and TensorFlow. The data was split into training, validation, and testing sets in the ratio 7:1:2, respectively. The deep learning algorithm chosen was the Recurrent Neural Network (RNN) because of its inherent feature extraction capabilities which is suitable for similar applications in previous ML PPI studies.19,23 The RNN comprises a deep learning structure that uses past information to improve the performance of the network on current and future inputs. 24 The training set of the data was used to train the network while the validation set was used to test the model’s performance at the end of each epoch (training cycle). Hyper-parameters (data weights, number of dense layers, activation functions, and loss functions) were adjusted to improve this validation accuracy and reduce validation loss until they did not change significantly after a given number of epochs. The model was fed with the tokenized sequences of 2 proteins, Protein_1_bait and Protein_1_prey, representing viral and host proteins, respectively, with their label (whether the protein pair interacts or not), as its inputs. The 2 proteins were processed in 2 separate branches of the network, where the features that describe the proteins were learned. Each branch comprised an embedding layer, a recurrent layer (with GRU units) and a fully connected layer to rearrange and recombine the information extracted by the 2 previous layers. The features that each extractor branch had computed were then merged into a single vector by concatenation, and the fully connected layer was used to combine the features of both proteins. The output was then interpreted as the probability of a sample (a protein pair) belonging to one of 2 classes (interaction, ⩾50% or no interaction < 50%). The final label was then assigned based on the score criterion. The parameters of the network were optimized by comparing the predicted label to the true label to improve the performance (Figure 2).

A schema summarizing the training of the neural network. The tokenized sequences in a given pair and their interaction label are fed into the neural network whose architecture is built to carry out feature extraction and then classification to obtain a model that can make predictions on new data.
Evaluation
After the training phase, the performance of the model was measured on the test data set using standard statistical metrics comprising balanced accuracy, precision, recall, specificity, Matthew’s Correlation Coefficient (MCC), F-1 score, and Area Under the Receiver Operating Characteristic Curve (ROC-AUC).25,26 These metrics are defined by the equations below:



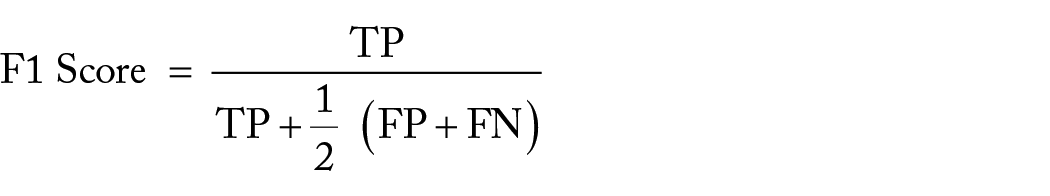

where TP, TN, FP, and FN represent the numbers of true-positive, true-negative, false-positive, and false-negative samples, respectively, based on the confusion matrix for classifications.
The model was further validated with 2 data sets: the Het-1A data set on HPV31 interactions, and a data set on HPV18 and host PPIs from Viruses.STRING. 17 The latter was used to investigate the predictive power of the trained model on HPV18 interactions despite having only trained on HPV31 interactions. The Het-1A data set of 84 positive interactions was pre-filtered against the combined HEK295 and C33A data set to eliminate common interactions and hence remove “familiar” contamination to it as external validation set. This dropped the positive interactions in Het-1A from 84 to 33. Tables 1 to 4 in the “Results” section summarize this.
Validation of performance of model using external data set showing the MiST and PPI model scores as well as the assigned labels.

Results for HPV18 and host protein interaction prediction by the model comprising confidence and PPI model scores.

HPV31 E6 and E7 interaction with AKT, IQGAP1, and MMP16 with PPI_model predictions and confidence scores.

HPV18 E6 and E7 interaction with AKT, IQGAP1, and MMP16 showing the confidence scores and PPI_model predictions.

Results
Evaluation of model performance
The values of the various metrics scored by the model on the test set (except for MCC) are provided (Figure 3). The MCC score was 0.7937, which indicated that the model was a good classifier.
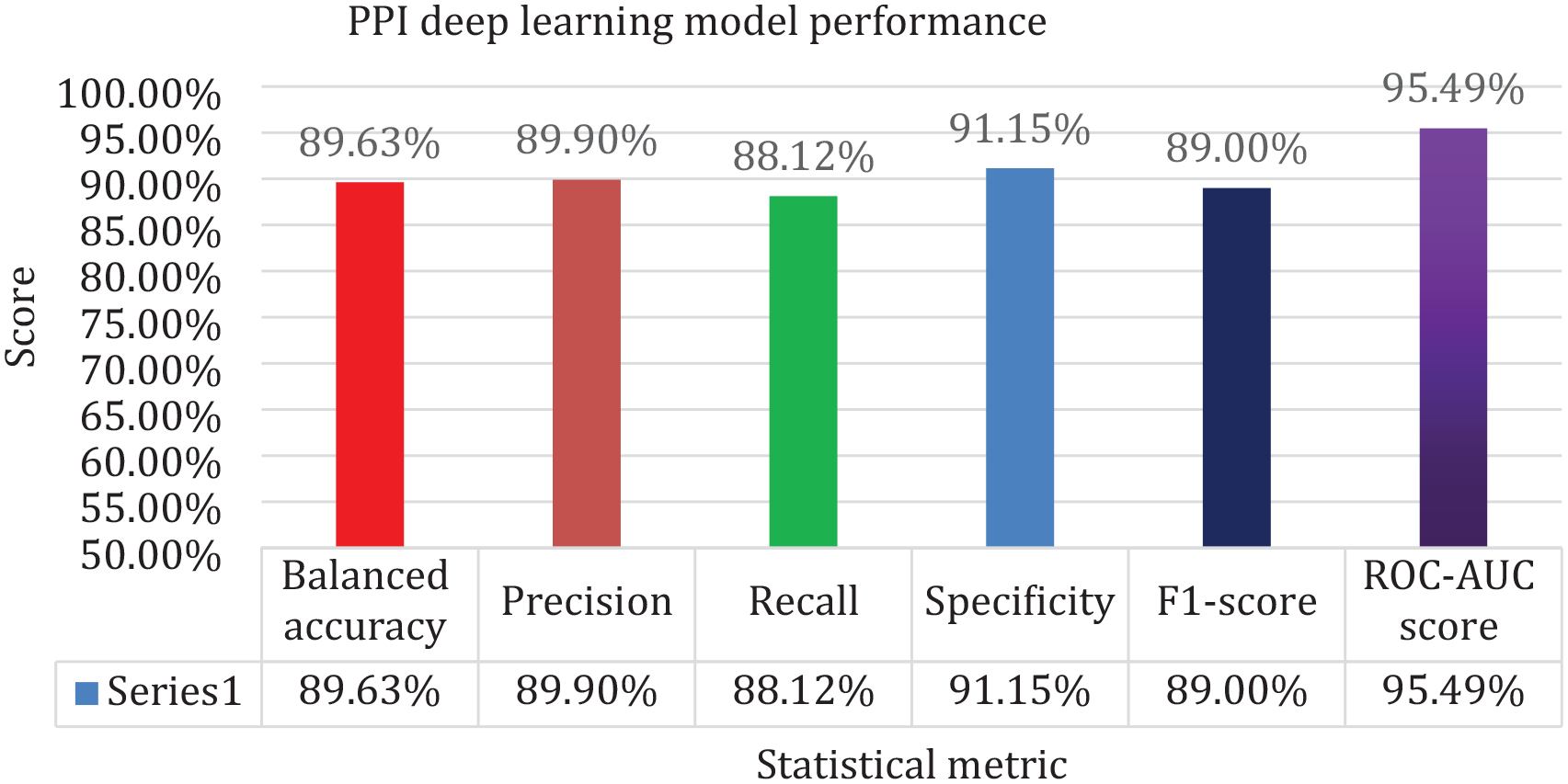
Model’s performance on the evaluation metrics. All the metrics scored above 88% indicating the model’s strong performance.
Generally, the model showed good performance, with all parameters showing performance above 88% which comparatively is not far off from the performance of models trained elsewhere.19,26
Validation of model
Out of a total of 33 HPV31 and host protein positive interactions from Het-1A used to validate the model, the model was able to correctly predict 25 as positive interactions (true positives), while 8 were predicted as negative interactions (false negatives). This gave the model a 75.75% recall on the Het-1A data set (Table 1).
The model was further tested to determine its ability to predict interaction between host proteins and proteins of a different HPV type (HPV18). A data set of 10 positive HPV18 and host PPIs was retrieved from Viruses.STRING database 17 and used to test the performance of the model. The model was able to correctly predict 7 out of the 10 positive interactions, giving it a recall of 70% on the HPV18 data set. The results for HPV18 and host protein interaction prediction by the model are shown in Table 2.
Model predictions
The model was then used to predict the possible interaction between HPV E6 and E7 and host oncoproteins AKT, IQGAP1, and MMP16. This was done for both HPV18 and HPV31, which gave quite similar results in predictions with only slight variations in probabilities. Table 3 presents the results for HPV31, while Table 4 shows those of HPV18.
Subsequently, HPV31 E6 and E7 were predicted to interact with AKT, while only HPV31 E7 was shown to interact with IQGAP1 and MMP16. Similarly, when the model was used to predict the possible interactions between HPV18 E6 and E7 and AKT, IQGAP1, and MMP16, the same predictions were made except with slight differences in confidence scores. This correlation points to shared similarities in the interactions HPV18 and HPV31 have with host oncoproteins AKT, IQGAP1, and MMP16. The resources including scripts used have been deposited in a GitHub repository (Access link: Supplementary).
Discussion
This study presents a model for predicting HPV and host PPIs using deep learning-based RNN algorithm. In this study, we set out to design a neural network to predict HPV and host protein interactions using the primary amino acid sequences. To achieve this, we utilized already available data, 16 thus circumventing the need for feature engineering associated with prediction problems, and then transformed the data to an RNN architecture selection problem. The resulting model is the first trained on this data set. The performance of our model was evaluated by criteria such as balanced accuracy, recall, precision, F-score, and MCC. Since PPI prediction is mostly aimed at correctly predicting the interacting protein pairs, the sensitivity and accuracy indicators were used to assess the model’s ability to predict positive data. Similarly, the MCC was used to evaluate the reliability and stability of the model when dealing with unbalanced data, and the receiver operating characteristic (ROC) was used to appraise the performance of a set of classification results while the area under ROC (AUC) was computed as an important evaluation indicator. 27
Our model showed good performance, with all the evaluation matrices ranging from 88% to 95%. When the performance of our model was compared to the performances of similar deep learning models,26,27 we noticed that the evaluation matrices of most of the models were slightly above the performance of our model (above 90%). For example, the performance of the deep learning model developed previously 27 on human, Helicobacter pylori, and Saccharomyces cerevisiae data sets had accuracy around 98% and sensitivity, precision, and MCC around 98.47%, 98.67%, and 97.19%, respectively. 27 Similarly, the performance of a deep neural network 26 on a combined data set comprising human, E. coli, Drosophila, C. elegans, and Mus musculus data sets achieved accuracy of 0.9941, recall of 0.9963, precision of 0.9915, F-score of 0.9939, and MCC of 0.9883. 23 The difference in performance observed between our model and other similar deep learning models could be due to the size of the data set used in the training of the models. For example, the data size used for the design of our model was approximately 1000 protein pairs, mainly because of paucity of data on HPV and host PPIs; while the data size of Wang et al, and Li et al, was approximately 70 000 and 60 000 protein pairs, respectively. Hence our data space may not have covered enough samples with intrinsic features. For instance, in Table 1, row 2, the interaction between HPV31L2_Sand SPTC1_HUMAN was the highest with an MiST score of 0.9901, and yet the model prediction scored 0.0787. This study used a 3-gram tokenization, which had been shown to produce robust outcomes. In future, once input data size increases considerably, the n-gram can be varied to evaluate its impact on the performance metrics. Similarly, the model can be optimized to learn features and increase its performance.
Other supervised ML models for PPI data sets have been trained to make predictions; however, they have been mainly based on the SVM algorithm. Five SVM models were trained on 16000 diverse PPI pairs 28 and the sequences were characterized by a conjoint triad descriptor and were used to train the models using a custom S-kernel function. They obtained accuracies > 82.75%, sensitivity values > 84.00% and precision scores > 82.75%. Similar models were trained, 29 however, the sequences were represented by feature vectors of consecutive amino acid triplets and the data set was for only HPV PPIs. 29 The average sensitivity was 77.8%, the average specificity was 85.4% and the average accuracy of 81.6%.
A vector representation of 3 major features of the sequences was used to train other SVM models. 30 The features consisted of the frequency difference of amino acid triplets, relative frequency of amino acid triplets, and amino acid composition. The models were trained with different combinations of the aforementioned features with the performance of 99.4% for sensitivity, 99.6% for specificity, 99.5% for accuracy, and an MCC of 0.989. Our PPI model developed which is a deep learning-based, performed better than some of these SVM models.
The ability of the model to predict proteins of other HPV types and host PPIs was also tested. This was achieved by testing the model’s performance on a data set of HPV18 and host PPIs, and a recall of about 70% was achieved; this is similar to the model’s performance on an evaluation data set of HPV31 and host PPIs (recall of 76%), suggesting that the model can be applied to predict other HPV types and host protein interaction effectively.
Finally, the model was then used to predict the possible interaction between E6 and E7 proteins of HPV18 and 31 and AKT, IQGAP1, and MMP16. From the results of the prediction, HPV E6 was found to only interact with AKT, while HPV E7 interacted with AKT, IQGAP1, and MMP16. The HPV E6 and E7 have been reported to initiate cancer by deregulating P53 and retinoblastoma protein (pRb), leading to unrestrained cell proliferation. 31 In addition, HPV E6/E7 expression has been reported to activate the PI3K signaling pathway and contribute to the amplification or mutation of the major components in this pathway. 32 The findings of the prediction suggest that HPV E6/E7 directly interact with some components of the P13K pathway (AKT, IQGAP1, and MMP16), and could explain the increased mutations and amplifications observed in components of the PI3K pathway in HPV-positive cancers. The results also suggest that E7 may be more involved in the deregulation of the PI3K pathway because of its ability to interact with more components of the pathway. Therefore, therapies targeting the disruption of E6/E7 and AKT, IQGAP1, and MMP16 interaction in HPV-associated cancers may be helpful in reducing the severity of these cancers. The deep learning model developed for prediction is primarily computational, and the predicted interactions must be corroborated using experimental assays.
Conclusions
We were able to develop for the first time a deep learning model for the prediction of HPV and host PPIs. The findings from the model’s prediction show that HPV E7 may interact more with components from the PI3K pathway (AKT, IQGAP1, and MMP16). These interactions should be a major driver in the deregulation of the pathway while E6 may contribute to activating AKT, leading to subsequent activation of other downstream oncogenes. These computationally elucidated interactions can be confirmed experimentally to provide insight into biomolecular mechanisms.
Footnotes
Acknowledgements
The authors specially thank Quaye Lab. Members of the Virology Lab, Department of Biochemistry, Cell and Molecular Biology, University of Ghana, for supporting the project.
Availability of Data and Materials
All data generated or analyzed during this study are included in this article.
Author Contributions
SKK, EAT, CAB, and OQ conceptualized and designed the work; SS, KA-M acquired and analyzed data or SKK, SS, and KA-M interpreted data; EAT and SS drafted the article; all authors revised it critically for important intellectual content; and all authors approved the version to be published.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Sheila Santa was supported by a WACCBIP-World Bank ACE PhD fellowship (WACCBIP + NCDs: Awandare)” and a DELTAS Africa grant (DEL-15-007: Awandare). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (107755/Z/15/Z: Awandare) and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust, or the UK government.