
Abstract
Atherosclerotic cardiovascular diseases (CVDs) are closely linked to factors such as familial hypercholesterolemia (FH), often caused by mutations in low-density lipoprotein receptor (LDLR) and apolipoprotein B (APOB). Through a comprehensive bioinformatic analysis, we identified novel LDLR and APOB mutations and their cardiovascular disease (CVD) implications, focusing on unique variants in the Vietnamese population. We used homology modeling to predict protein structures; in addition, through protein-protein molecular docking, we assessed how these mutations affect binding affinities. We identified 10 novel binding residues exclusive to the wild-type and precursor LDLR isoforms, including ASP-47, GLY-48, and GLU-51. Analyses of 154 complexes revealed 5 isoforms with low binding affinities and notable hydrogen-bonding interactions—APOB (Arg3527Trp)-LDLR (Cys318Arg), APOB (His3583Leu)-LDLR (Cys104Tyr), APOB wild-LDLR (Glu228Lys), APOB (Phe2469Cys)-LDLR (Glu288Lys), and APOB wild-LDLR (Ser130Ter). These results suggest strong and potentially detrimental interactions among these proteins. Furthermore, they highlight the molecular mechanisms underlying CVD development, reveal potential therapeutic targets, enhance our understanding of genetic variations, and could guide FH research.
Introduction
Familial hypercholesterolemia (FH) is an inherited dyslipidemia characterized by increased levels of low-density lipoprotein-cholesterol (LDL-C), primarily attributed to mutations in low-density lipoprotein receptor (LDLR) and apolipoprotein B 100 (APOB). 1 In a recent meta-analysis, the prevalence of FH was 1 in 313 individuals, varying across populations. 2 However, several knowledge gaps persist in understanding FH prevalence and specific mutations associated with FH in the Asian populations, including the Vietnamese population.
Genetic aberrations in LDLR and APOB contribute to the early onset of atherosclerosis and pose a substantial risk of cardiovascular diseases (CVDs). The LDLR, a multifaceted transmembrane protein, plays a pivotal role in LDL-C clearance from the circulation. Mutations in LDLR can disrupt LDL-C uptake, leading to increased plasma cholesterol levels. 3 The complex interplay between LDLR and APOB-100 is crucial for cholesterol metabolism. The APOB-100 serves as an LDL-C carrier and interacts with the ligand-binding domain of LDLR to facilitate the internalization and processing of LDL particles. The LDLR binds to LDL particles through interactions with APOB-100. This binding triggers receptor-mediated endocytosis, wherein the cell membrane invaginates to form a vesicle that internalizes the LDL-LDLR complex. The vesicle fuses with lysosomes, thereby degrading LDL to release cholesterol for cellular use, whereas LDLR is recycled to the cell surface, assisting in the regulation of cholesterol levels. This complex interplay between LDLR and APOB-100 is crucial for cholesterol metabolism. The APOB-100 serves as an LDL-C carrier and interacts with the ligand-binding domain of LDLR to facilitate the internalization and processing of LDL particles. Mutations in APOB that impede the binding of the encoded protein to LDLR have been implicated in FH pathogenesis. There are multiple functional classes of mutations that prevent the synthesis of LDLR (class 1), hinder its transport to the cell surface (class 2), impair its ability to bind LDL (class 3), block internalization of the LDL-LDLR complex (class 4), and prevent receptor recycling after LDL degradation (class 5). Each class of mutation disrupts a different step in the life cycle of the receptor, leading to elevated LDL cholesterol levels, often contributing to FH development. Therefore, a comprehensive understanding of the genetic basis of LDLR and APOB, particularly functional implications of mutations, is essential for elucidating the mechanisms underlying FH development. 4 Furthermore, determining the roles of proteins encoded by these genes may provide valuable insights into the enhanced susceptibility of individuals with these mutations to atherosclerotic CVDs.
Recent studies have reported molecular modeling of LDLR5,6; however, the synergistic effects of LDLR and APOB mutations on FH remain unknown. Moreover, the molecular mechanisms underlying the binding of APOB-100 to LDL-C via LDLR remain inadequately elucidated. 7 Therefore, a comprehensive approach, including genetic analysis, structural modeling, binding site identification, molecular docking, and residue-wise interaction analysis, is required to elucidate the roles of LDLR and APOB-100 proteins in FH pathogenesis. To address these gaps in the literature, in the present study, we obtained mechanistic insights into cholesterol metabolism at the molecular level. We elucidated genetic variations and structural complexities associated with FH mutations in the Vietnamese population, which shares genetic characteristics with other East and Southeast Asian populations, 8 using the FH genetic database in the Vietnam Familial Hypercholesterolemia (VINAFH) Program 9 to analyze APOB-100 and LDLR proteins. The findings hold potential to revolutionize therapeutic approaches for CVD by providing new insights into the underlying genetic factors and molecular interactions that could be targeted for reducing lipid levels.
Materials and Methods
Computational analysis and genetic data collection
We employed a multifaceted approach, commencing with the identification of LDLR and APOB mutations through comprehensive data mining and prediction tools. A graphical overview of the study process is presented in Figure 1. To acquire information on single-nucleotide polymorphisms (SNPs) of APOB and LDLR, along with their pathogenicity and mutation positions, the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and dbSNP databases were used. For further verification of mutations, 2 prediction tools, PolyPhen2 and Mutation Taster, were employed. Homo sapiens LDLR and APOB sequences were retrieved from the UniProtKB database (https://www.uniprot.org/uniprotkb). Moreover, in silico mutagenesis was performed to replace wild-type residues with respective mutations in the LDLR and APOB-100 sequences using the Bio.seq module and Bio.seqIO, the standard Sequence Input/Output interface for BioPython (version 1.43).

Graphical representation of the study overview.
All variants were detected in FH index cases from the Vietnamese population as part of an extended and developed database of the VINAFH program. 9 The LDLR and APOB-100 amino acid sequences were retrieved from the UniProtKB database for further analysis. The inclusion criteria for the data of patients with FH included positive LDLR and/or APOB mutations. These index cases exhibited FH phenotypes according to the Dutch Lipid Clinic Network Score criteria for adults and the Wiegman criteria for children aged <18 years. 10 Genetic testing was performed for all index cases to determine the genotype, including homozygous FH, compound heterozygous FH, and simple heterozygous FH. Various genetic testing methods, including next-generation sequencing, massively parallel sequencing, multiplex ligation-dependent probe amplification, Sanger sequencing for LDLR,9,11 and whole-exome sequencing, were employed. In this study, we excluded computerizable construction and analysis for LDLR mutations indicative of complete loss or absence of functional LDLR (null LDLR mutations) and APOB mutations associated with familial hypobetalipoproteinemia.
This study was conducted according to the guidelines of the Declaration of Helsinki and was approved by the Ethics Committee of Hanoi Medical University (approval number: 39/GCN-HĐĐĐNCYSH-ĐHYHN, approval date: April 19, 2021). Informed consent for using genetic data was obtained from all patients with FH or from the parents or legal representatives of those aged <18 years.
Homology modeling for low-density lipoprotein receptor and apolipoprotein B proteins
Homology modeling was performed on the LDLR structures from AlphaFold (P01130) and OmegaFold, LDLR isoform-3 precursor, LDLR isoform-4 precursor, and APOB-100 (P04114) wild-type and mutant structures. The following 2 protein structure databases for LDLR were accessed: AlphaFold, an open-source extensive database of high-accuracy protein structure predictions (https://alphafold.ebi.ac.uk), and OmegaFold, a computational method for successfully predicting high-resolution protein structures from a single primary sequence. MODELLER (V10.4) was used to employ homology modeling techniques for predicting the structures of mutated LDLR proteins. This approach involved incorporating mutations into the sequences generated by the AlphaFold and OmegaFold databases through mutagenesis and investigating the effect of specific residues at various positions within the protein sequence. Moreover, homology modeling was performed to determine structures of the LDLR isoform 3 and 4 precursors. Ten LDLR models were generated for each mutant, and the best model was selected based on the Discrete Optimized Protein Energy score.
Three templates were predicted based on the length of the APOB-100 sequence using Robetta, 12 an automated protein structure prediction server using the Rosetta fragment insertion method. This method helps integrate template-based and de novo structure prediction methods to produce high-quality models covering every submitted sequence residue. 13 These templates were combined using multiple-template homology modeling to obtain a complete predicted structure. Multiple-template modeling techniques were employed using MODELLER (V10.4) to predict the complete structure of APOB-100. Ten models were generated, and the best model was selected based on the molpdf score. Furthermore, the mutations in APOB were incorporated using MODELLER (V10.4). The resulting structures were evaluated using various tools, including the structural assessment web server SAVES v6.0 (https://saves.mbi.ucla.edu/) for ERRAT (score >70%), as well as ProSA (<−1 and within the range of the experimental structures) (https://prosa.services.came.sbg.ac.at/prosa.php), Ramachandran Plot Server (>90% most favored regions, <2% disallowed regions) (https://zlab.umassmed.edu/bu/rama/), and QMEAN (>−2) (https://swissmodel.expasy.org/qmean/).
Identification of binding sites
The relevant binding sites were identified via a thorough literature review using the PubMed, ScienceDirect, and Google Scholar databases. This review was instrumental in consolidating existing knowledge of the binding interfaces between APOB-100 and LDLR. Identifying the precise sites of protein-ligand or protein-protein interactions is imperative for elucidating protein functionalities and exploring their putative roles. The computational tool GRaSP was employed for in silico prediction of protein-protein binding sites. The GRaSP considers the characteristics of glycoproteins, radii, and atomic solvation parameters to identify binding sites between LDLR and APOB-100. 14
Molecular docking, residue-wise protein-protein interaction analysis, and visualization
To analyze molecular interactions, a 3-dimensional model of protein complexes obtained from HDOCK was examined. 15 These complexes included various combinations, including APOB wild-type with LDLR wild-type, APOB wild-type with LDLR mutants, LDLR wild-type with APOB mutants, LDLR mutants with APOB mutants, LDLR isoform-3 precursor wild-type with APOB wild-type, APOB wild-type with LDLR isoform-3 precursor mutant, LDLR isoform-3 precursor wild-type with APOB mutants, LDLR isoform-3 precursor mutant with APOB mutants, LDLR isoform-4 precursor wild-type with APOB wild-type, APOB wild-type with LDLR isoform-4 precursor mutant, LDLR isoform-4 precursor wild-type with APOB mutants, and LDLR isoform-4 precursor mutant with APOB mutants obtained from HDOCK. 15 The HDOCK uses a hybrid algorithm that combines template-based and template-free docking methods. We employed the PDBSUM server (http://www.ebi.ac.uk/pdbsum), a web-based database offering concise and visually rich summaries of macromolecular structures from the Protein Data Bank (PDB). This server helped predict receptor-ligand interactions based on information such as amino acid sequences and PDB structures. The PDBSUM generates 100 theoretical models depicting potential protein-protein (peptide) interactions and evaluates their docking energy scores. Model 1 from each category was selected based on the highest confidence score and lowest binding energy, which provided images of structures, secondary structures, detailed structural analyses, quality assessments, and schematic illustrations of protein-ligand and protein-DNA interactions. Using this tool, bonded and nonbonded interacting residues of protein-protein interactions were analyzed. All complex structures were visualized in PyMol (The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC, New York, USA) to present stick and surface representations, highlight interacting residues, and display polar interactions between docked protein complexes.
Results
Genetic variations in low-density lipoprotein receptor and apolipoprotein B in the Vietnamese population
We identified 20 variants in LDLR (Cys104Tyr, Ser130Ter, Asp168Tyr, Cys222Arg, Asp227Glu, Glu228Lys, Arg257Trp, Glu288Lys, Cys318Arg, Pro476Arg, His583Tyr, Ser586Pro, Asp589Asn, Ser648Pro, Gln660Ter, Ala684Thr, Val797Met, Val806fs, Asp843Glufs*86, and Gly844Ser) and 6 variants in APOB (Arg1386Trp, Phe2469Cys, His3583Leu, Arg4297His, Trp3153Arg, and Arg3527Trp) specific to the Vietnamese population with FH. These variants were analyzed to investigate interactions between mutations in LDLR and those in APOB-100. In silico prediction tools, including PolyPhen2 and MutationTaster, helped identify clinically significant mutations (Table 1).
Retrieved LDLR and APOB variants with the corresponding coding sequence, exon, amino acid substitutions, and domain region.

APOB, apolipoprotein B.
LDLR, low-density lipoprotein receptor; VINAFH, Vietnam familial hypercholesterolemia program.
Structure prediction and evaluation of wild-type and mutant models
Evaluation of the AlphaFold and OmegaFold structures of LDLR revealed superior quality assessment scores for the AlphaFold structure compared with those for the OmegaFold structure across all evaluation servers (Supplementary Tables S1 and S2). This result suggests that the LDLR AlphaFold structures are more suitable for further analysis than the OmegaFold structures.
Identification and characterization of binding sites
We focused on the binding sites associated with APOB-100, spanning residues 3000 to 3700. Specifically, 2 short heparin-binding sites (regions 3134-3209 and 3356-3489), rich in basic amino acids, contributed significantly to the heparin-binding properties of these sites. Individually or in combination, these sites may constitute the receptor-binding domain of APOB-100. Regions 3143 to 3155 and 3356 to 3368 play crucial roles in various biological processes and interactions involving APOB-100, facilitating interaction or binding between APOB-100 and LDLR (Supplementary Table S3).
The APOB-100 sequencing revealed arginine-rich and lysine-rich regions at residues 3147 to 3157 and 3359 to 3367, respectively, which were identified as potential LDLR-binding sites. Previous findings have indicated structural similarities between the 3345 and 3381 regions in APOB-100, known for binding to LDLR. Experiments involving a synthetic peptide corresponding to residues 3345 to 3381 in APOB-100 demonstrated the importance of this region in the binding and internalization of LDL particles by LDLR. The Law and Scott’s comparative analysis of APOB-100 sequences across 7 species highlighted residues spanning 3359 to 3367 within APOB-100 as the primary site involved in receptor interaction (Supplementary Table S3). The advanced GRaSP tool facilitated binding residue identification in different LDLR variants—ASP47, GLY48, GLU51, ASP57, and GLU58 for LDLR wild-type; ASP88, ASP90, ASP92, ASP98, and GLU99 for LDLR isoform-3 precursor; and ASP36, ASP47, GLY48, ALA50, GLU51, ASP54, ASP57, and GLU58 for LDLR isoform-4 precursor. These residues, each with significant confidence scores, play crucial roles in facilitating the interaction or binding between APOB-100 and LDLR. Thereafter, we constructed 154 APOB-LDLR complexes. The surface view of each complex and combined surface view of these complexes are presented in Supplementary Figures S1 to S154.
Comparative analysis of docking complexes: wild-type vs mutants
Wild-type apolipoprotein B-low-density lipoprotein receptor complexes with wild-type apolipoprotein B-low-density lipoprotein receptor mutant complexes
The molecular docking analysis of the APOB wild-type with LDLR wild-type demonstrated a binding affinity of −345.38. Conversely, the APOB wild-type with LDLR mutants (Cys104Tyr, Ser130Ter, Asp168Tyr, Cys222Arg, Glu228Lys, Arg257Trp, Asp227Glu, Glu288Lys, Cys318Arg, Pro476Arg, His583Tyr, Ser586Pro, Asp589Asn, Ser648Pro, Gln660Ter, Ala684Thr, Asp843Glufs*86, Val797Met, Val806fs, and Gly844Ser) exhibited an average binding affinity of −338.41 kcal/mol, which is slightly weaker than that of the wild-type APOB-LDLR complex (Table 2).
Docking score of the APOB-LDLR complexes.

On comparing the wild-type APOB-LDLR complex with the wild-type APOB-LDLR mutant complex, we observed that the complexes APOB wild-type with LDLR (Gly844ser), APOB wild-type with LDLR (Ala684Thr), APOB wild-type with LDLR (Glu228Lys), APOB wild-type with LDLR (Ser130Ter), and APOB wild-type with LDLR (His583Tyr) displayed the lowest binding affinities of −315.25, −316.12, −321.03, −324.56, and −328.99 kcal/mol, respectively (Figure 2). These values are higher (less negative) than the value of the wild-type complex, indicating weaker binding. On the contrary, certain mutant complexes, such as the wild-type APOB with LDLR (Cys222Arg, Pro476Arg, Gln660Ter, Ser586Pro, and Ser648Pro), exhibited stronger binding affinities of −379.11, −357.57, −356.53, −352.93, and −348.85 kcal/mol, respectively, which are lower (more negative) than the value of the wild-type complex, indicating stronger binding (Table 2).
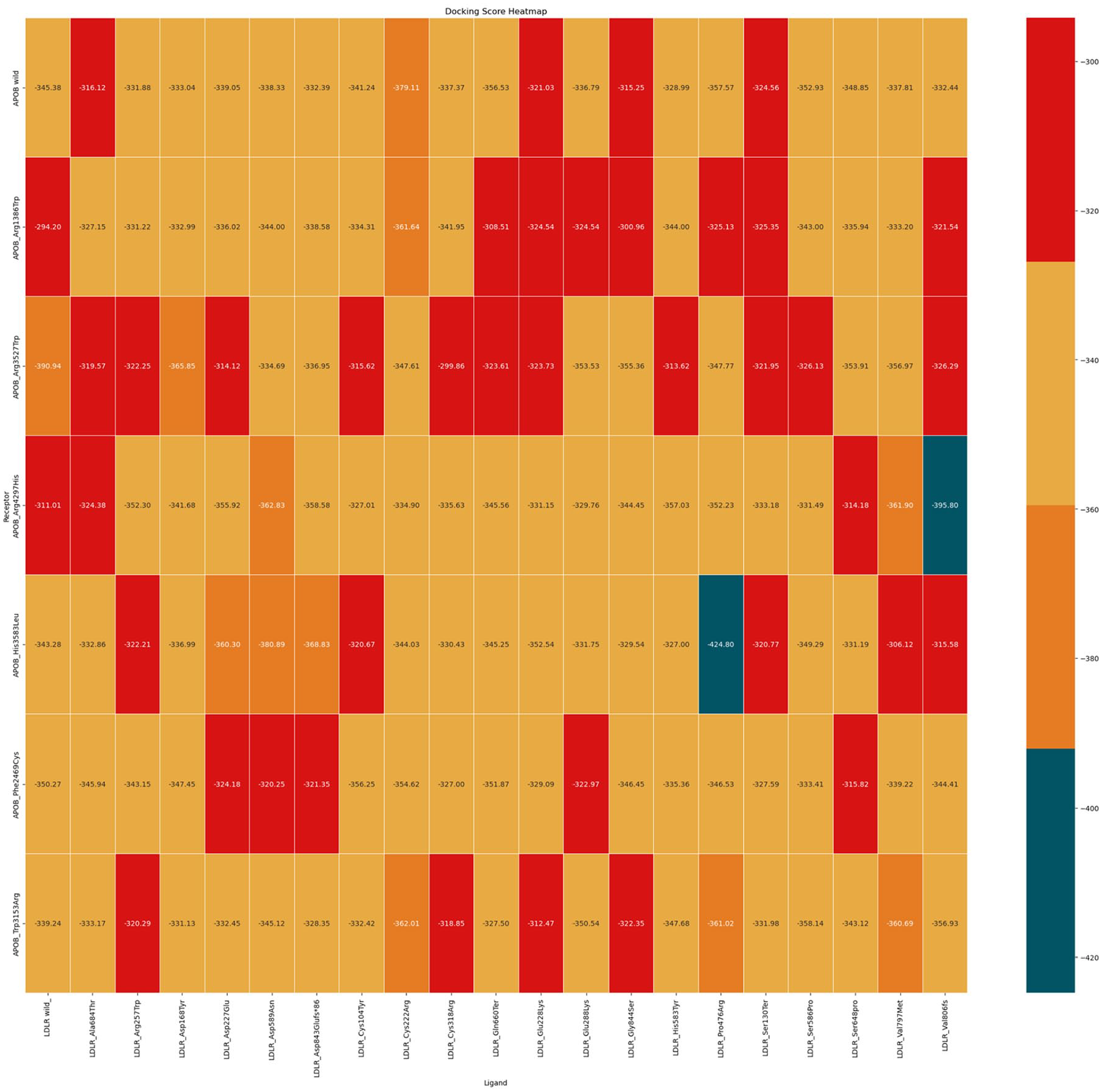
Heatmap showing the binding affinities of complexes using colors. Low binding affinity complexes are represented in red, whereas highest binding affinity complexes are represented in blue.
The APOB mutants (Arg1386Trp, Phe2469Cys, His3583Leu, Arg4297His, Arg3527Trp, and Trp3153Arg) with LDLR wild-type exhibited an average binding affinity of −339.19 when compared with their wild-type APOB-LDLR complex (Table 2). When comparing the wild-type APOB-LDLR complex with the mutant APOB-LDLR wild-type complex, we observed that the complexes of LDLR-wild-type with APOB (Arg1386Trp), LDLR-wild-type with APOB (His3583Leu), and LDLR-wild-type with APOB (Arg4297His) displayed weaker binding affinities, ranging from −343.28 to −294.2 kcal/mol, than the wild-type APOB-DLR complex. Specifically, the complex LDLR wild-type with APOB (Arg1386Trp) exhibited the weakest binding affinity of −294.2 kcal/mol. In contrast, the complex LDLR wild-type with APOB (Arg3527Trp) showed the strongest binding affinity of −390.94 kcal/mol, which is significantly stronger than that of the wild-type complex (Table 2). The wild-type APOB-LDLR docking complex and its interaction sites are illustrated in Figures 3 and 4.

APOB-LDLR interaction sites. A docked complex of wild-type APOB-LDLR (showing interaction sites) is shown in (A). The closest poses of APOB and LDLR interactions are shown in (B) and (C). Deep salmon shade shows APOB, and teal represents LDLR; APOB-binding residues are shown in red, and LDLR-binding residues are displayed in orange. The interacting residues and respective bonds are highlighted in (D). Salt bridges are shown in red, and disulfide bonds are displayed in yellow; blue represents hydrogen bonds, and orange shows nonbonded contacts.
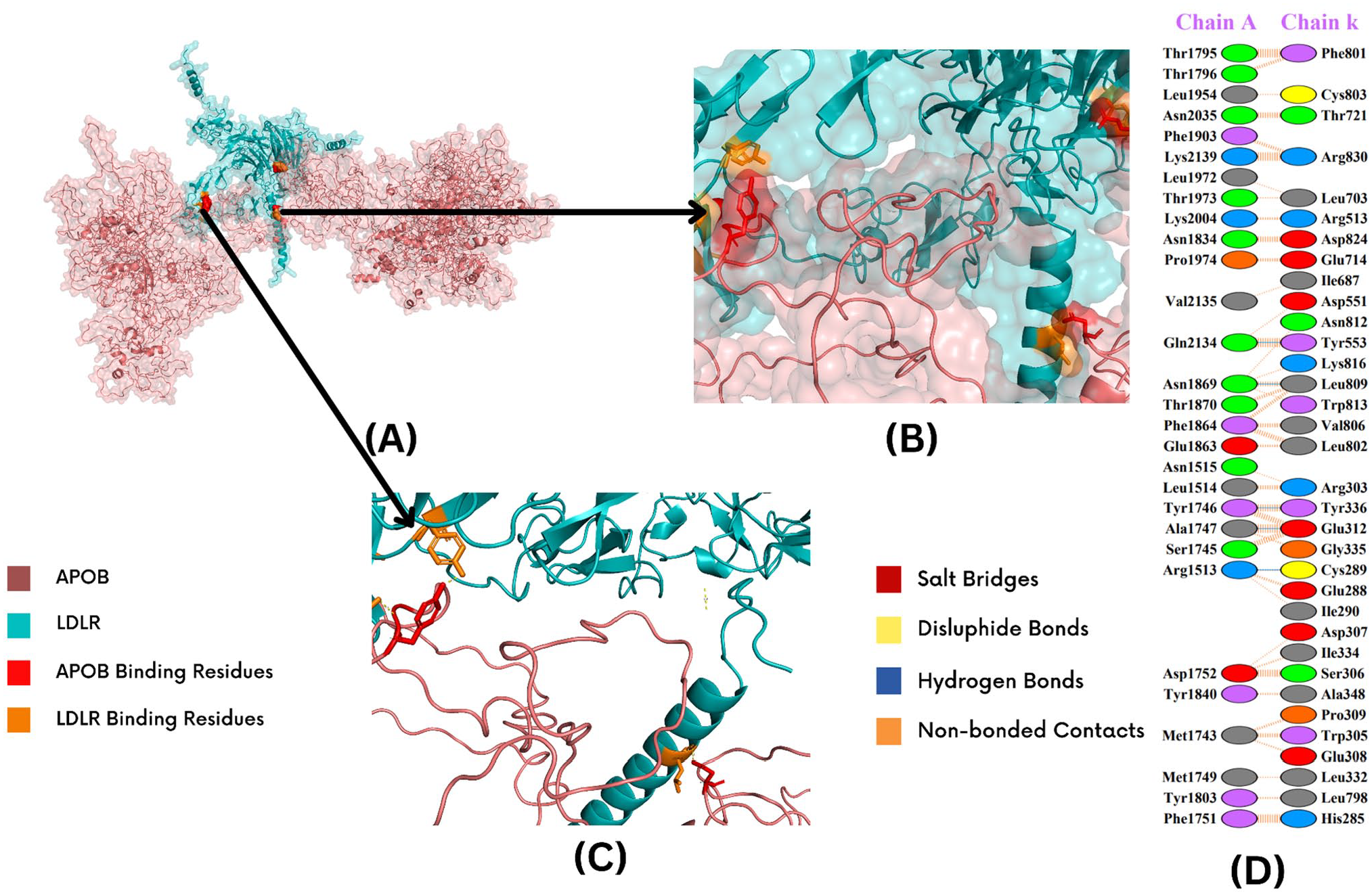
LDLR-APOB (Arg1386Trp) interaction sites. Docked complex of LDLR-APOB (Arg1386Trp), showing interaction sites is shown in (A). The closest poses of APOB and LDLR interactions are shown in (B) and (C). Deep salmon shade shows the APOB, and teal represents the LDLR; APOB-binding residues are shown in red, and LDLR-binding residues are displayed in orange. The interacting residues and respective bonds are highlighted in (D). Salt bridges are shown in red, and disulfide bonds are shown in yellow; blue represents the hydrogen bonds, and orange depicts the nonbonded contacts.
Wild-type apolipoprotein B-low-density lipoprotein receptor complex with mutant apolipoprotein B-low-density lipoprotein receptor complexes
The molecular docking analysis of the APOB wild-LDLR wild complex exhibited a binding affinity of −345.38 kcal/mol. A comparison of the wild-type APOB-LDLR with mutant APOB-LDLR complexes revealed that certain mutant complexes displayed significantly reduced binding affinities. Specifically, the complexes APOB (Arg3527Trp) with LDLR (His583Tyr), APOB (Trp3153Arg) with LDLR (Glu228Lys), APOB (Arg1386Trp) with LDLR (Gln660Ter), APOB (His3583Leu) with LDLR (Val797Met), APOB (Arg1386Trp) with LDLR (Gly844Ser), and APOB (Arg3527Trp) with LDLR (Cys318Arg) exhibited the weakest binding affinities of −313.62, −312.47, −308.51, −306.12, −300.96, and −299.86 kcal/mol, respectively (Supplementary Table S4). In contrast, several complexes demonstrated significantly stronger binding affinities than the wild-type complex. The complexes APOB (His3583Leu) with LDLR (Pro476Arg), APOB (Arg4297His) with LDLR (Val806fs), and APOB (His3583Leu) with LDLR (Asp589Asn) exhibited the strongest binding affinities of −424.8, −395.8, and −380.89 kcal/mol, respectively.
Wild-type apolipoprotein B-low-density lipoprotein receptor isoform 3 precursor complexes with mutant apolipoprotein B-low-density lipoprotein receptor wild-type complexes
The molecular docking analysis of the wild-type APOB-LDLR isoform 3 precursor complex exhibited a binding affinity of −328.75 kcal/mol. In comparison, the wild-type APOB-LDLR isoform 3 precursor complexes with mutant APOB-LDLR wild-type complexes demonstrated an average binding affinity of −313.61 kcal/mol (Supplementary Table S5). Compared with the wild-type complex, the wild-type LDLR isoform 3 precursors with APOB (His3583Leu), APOB (Phe2469Cys), wild-type APOB (Arg4297His), and APOB (Arg1386Trp) complexes exhibited low binding affinities, indicating weak binding.
Wild-type apolipoprotein B-low-density lipoprotein receptor isoform 4 precursor complexes with mutant apolipoprotein B-low-density lipoprotein receptor wild-type complexes
The molecular docking analysis of the wild-type APOB-LDLR isoform 4 precursor complex showed a binding affinity of −353.17. In comparison, the wild-type APOB-LDLR isoform 4 precursor complexes with mutant APOB demonstrated an average binding affinity of −340.64 (Supplementary Table S6). Compared with the wild-type complex, the complexes wild-type LDLR isoform 4 precursors with APOB (Phe2469Cys), wild-type LDLR isoform 4 precursor with APOB (His3583Leu), and wild-type LDLR isoform 4 precursor with APOB (Arg4297His) exhibited the lowest binding affinities of −317.47, −316.27, and −307.64 kcal/mol, respectively, indicating weaker binding. In contrast, the complex of wild-type LDLR isoform 4 precursor with APOB (Trp3153Arg) showed the highest binding affinity of −363.56 kcal/mol, indicating stronger binding than that for the wild-type complex.
Residue-wise protein-protein interaction analysis
The residue-wise protein-protein interaction analysis performed using PDBsum1 revealed diverse protein-protein interactions along with residues and positions in all the complexes (Supplementary Table S7). When analyzing 154 complexes, we observed that 26 complexes exhibited higher hydrogen-bonding interactions in the range of 10 to 18. Furthermore, repeated residue interactions across all complexes were analyzed to identify consistent patterns in the manner residues interact with each other, regardless of whether those residues belonged to wild-type or mutant structures. These hotspot-interacting residues on the surfaces of LDLR and APOB are presented in Figure 5.

Visual representation of interacting hotspot residues of APOB and LDLR. (A) Deep salmon shade represents the APOB protein structure. Red represents the interacting hotspot residues of APOB, whereas blue represents the mutated residues. (B) Teal represents the APOB protein structure. Orange represents the interacting hotspot residues of APOB, whereas hot pink represents the mutated residues.
Finally, the interactions among the complexes at mutation sites were examined to comprehend the mechanisms by which specific genetic mutations might affect the function and stability of proteins. The hydrogen-bonded interaction ARG (318)-THR (1783) was identified within the mutated arginine residue of LDLR and the threonine residue of APOB. This interaction occurred specifically in the APOB (His3583Leu)-LDLR (Cys318Arg) complex.
The hydrogen-bonded interaction GLU (288)-THR (53) was observed in mutated complexes involving APOB (His3583Leu)-LDLR (Cys318Arg), APOB (His3583Leu)-LDLR (Ser648Pro), and APOB (Arg1386Trp)-LDLR (Ser648Pro), whereas TYR (168)-SER (2901) was observed to involve the wild-type tyrosine residue of LDLR isoform-4 precursor and serine of APOB. This interaction included the wild-type glutamic acid residue in the LDLR at the mutated site. The ASP (843)-ARG (4179) was identified in mutated APOB (Arg4297His)-LDLR (Asp168Tyr) and APOB (Arg3527Trp)-LDLR (Asp168Tyr), whereas an ASP (843)-TYR (2549) interaction was observed in APOB wild-LDLR (Asp168Tyr). These interactions included the wild-type aspartic acid residue in LDLR at the mutated position. The interactions ARG (3527)-ASP (54) and ARG (3527)-SER (157) involved the APOB wild-type arginine residue within APOB (Arg4297His)-LDLR (Ser130Ter) and APOB wild-LDLR (Gln660Ter).
Discussion
In this study, we comprehensively analyzed the interplay of LDLR-APOB modeling for the first time; we identified 6 APOB and 20 LDLR genetic variants in the Vietnamese population and elucidated the complex mechanisms underlying CVD development. Homology modeling was employed to predict the mutant structure of LDLR obtained from OmegaFold and AlphaFold. Given the unavailability of the complete structure of APOB-100, 3 templates were generated using Robetta. These templates were merged via multiple-template homology modeling to construct a complete predicted structure. Structural evaluation employing ERRAT, ProSA, QMEAN, and the Ramachandran plot server elucidated the superiority of AlphaFold models of LDLR over those obtained from OmegaFold, corroborating their suitability for thorough analyses. This rigorous structural assessment ensures the reliability of subsequent research on the effect of genetic variants on protein interactions and disease progression.
We identified pivotal binding sites associated with LDLR and APOB-100, focusing on their contributions to protein function and interactions. Although 9 APOB-100 binding sites have been previously identified within specific residues in the LDLR, our significant breakthrough lies in the discovery of previously unidentified binding residues, including ASP-47, GLY-48, GLU-51, ASP-57, GLU-58, ASP-88, ASP-90, ASP-92, ASP-98, GLU-99, ASP-36, ASP-47, GLY-48, ALA-50, GLU-51, ASP-54, ASP-57, and GLU-58, exclusive to the wild-type and precursor isoforms of LDLR. The identification of these novel binding sites could significantly improve our comprehension of LDLR function, ultimately enriching our knowledge of cholesterol metabolism and its implications for cardiovascular health. 16
The molecular docking analysis within the context of wild-type and mutant complexes within the APOB-LDLR system unveiled crucial insights into potential disease-related interactions. Particularly, the complexes wherein wild-type APOB bound with LDLR mutants and mutant APOB bound with mutant LDLR, including APOB wild-type with LDLR (Gly844ser), APOB wild-type with LDLR (Ala684Thr), APOB (Arg3527Trp) with LDLR (His583Tyr), APOB (Trp3153Arg) with LDLR (Glu228Lys), APOB (Arg1386Trp) with LDLR (Gln660Ter), APOB (His3583Leu) with LDLR (Val797Met), APOB (Arg1386Trp) with LDLR (Gly844Ser), and APOB (Arg3527Trp) with LDLR (Cys318Arg), exhibited the lowest binding affinities. These low binding affinities within the APOB-LDLR complexes can markedly disrupt the interactions responsible for maintaining cholesterol homeostasis. 17 When binding affinities are compromised, the normal processes of LDL particle internalization and cholesterol utilization are impeded, resulting in LDL-C accumulation in the bloodstream.17,18 This disruption in cholesterol homeostasis constitutes a crucial factor in the onset of CVDs. Elevated circulating LDL-C levels resulting from these distorted interactions create a conducive milieu for atherosclerosis and CVD development, highlighting the intricate relationship among novel genetic mutations, protein binding, and their profound ramifications in cardiovascular health.19,20
Conversely, complexes involving LDLR wild-type and APOB mutants exhibited a wide range of binding affinities for the wild-type APOB-LDLR complex. The complex involving LDLR wild-type with APOB (Arg1386Trp) displayed the lowest binding affinity. The APOB-LDLR isoform-4 precursor complex with wild-type APOB and the LDLR mutant, and mutant APOB-LDLR wild-type showed notably lower binding affinities, especially in the cases of APOB wild-type with LDLR (Ser418Pro), wild-type LDLR isoform-4 precursor with APOB (Phe2469Cys), wild-type LDLR isoform-4 precursor with APOB (His3583Leu), and wild-type LDLR isoform-4 precursor with APOB (Arg4297His). Detailed analysis of the protein-protein interactions within the APOB and LDLR complexes provided crucial insights into the potential consequences of novel genetic mutations, providing insights into their effects on protein function, disease development, and stability. Our findings align with those of previous studies, wherein researchers have shown how mutations in APOB and LDLR cause defects in the functions of both proteins; furthermore, a few studies have reported mutations outside the main binding region of APOB-100.21 -24
Protein-protein interactions are pivotal for mediating metabolic and signaling pathways, governing cellular processes, and the overall functioning of an entire organism. 25 Diseases frequently arise from mutations that disrupt these interactions, affecting the binding interfaces or inducing biochemically dysfunctional allosteric changes in proteins. 26 We identified a spectrum of hydrogen-bonded interactions among different combinations of APOB and LDLR mutant and wild-type complexes, highlighting the complexity of these interactions. These hydrogen bonds are crucial for maintaining stable protein-protein interactions during different phases of complex protein and cellular functions. 27 Studies have reported that reduction in the number of hydrogen bonds can cause defects in interactions among key proteins, leading to detrimental protein functions.28,29
A complex with the lowest binding affinity and the highest number of interactions can exert a significant influence on the severity and pathogenicity of diseases. 30 In the present study on LDLR and APOB proteins, complexes, including APOB (Arg3527Trp) with LDLR (Cys318Arg), APOB (His3583Leu) with LDLR (Cys104Tyr), APOB wild-type with LDLR (Glu228Lys), APOB (Phe2469Cys) with LDLR (Glu288Lys), and APOB wild-type with LDLR (Ser130Ter), demonstrated a stronger and potentially more deleterious interaction between these proteins, which might be linked to the development and progression of CVDs. Several mutants of LDLR (such as Gly844Ser and Ala684Thr) exhibited weaker binding affinities than the wild-type complex. These weaker binding interactions likely indicate reduced efficiency of LDLR-mediated clearance of LDL particles, as a lower binding affinity between APOB-100 and LDLR could result in decreased receptor-ligand interaction. Consequently, this would impair the ability of LDLR to internalize LDL particles from the bloodstream, leading to elevated LDL cholesterol levels, a hallmark of FH. Such reduced binding affinities may directly correlate with the development of FH by contributing to cholesterol buildup and subsequent atherosclerosis due to poor LDL clearance. On the contrary, the stronger binding affinities observed in complexes such as Cys222Arg and Pro476Arg indicate abnormal interactions between APOB-100 and LDLR, potentially leading to improper receptor recycling or internalization of LDL particles. Although these stronger affinities might seem advantageous, they could disrupt normal receptor dynamics, contributing to the development of FH by causing cholesterol dysregulation and accumulation in the bloodstream. A low binding affinity implies a tight connection between molecules, possibly leading to increased disease severity and pathogenicity, as these interactions can disrupt normal cellular processes and contribute to the clinical manifestations of the disease. 30
The examination of repeated residue interactions across all complexes revealed consistent patterns. Specific residue pairs, including GLU (3957)-ARG (494), ARG (4446)-THR (21), ASN (4447)-THR (21), and TYR (443)-ARG (303), recurred in various complexes. These repeated interactions highlight the significance of specific binding sites and their role in modulating protein-protein interactions in the APOB-100 and LDLR complexes. Hydrogen-bonded interactions involving mutant residues were observed at mutation sites ARG (318)-THR (1783) in the APOB (His3583Leu)-LDLR (Cys318Arg) complex, providing crucial insights into the novelty of the mutation. Notably, some LDLR mutations affect the APOB-LDLR interaction causing FH and are classified as classes 3 and 4. Therefore, in this study, we confirmed that the LDLR mutations are of classes 3 and 4 that weaken the interaction between APOB-100 and LDLR and that other mutations do not affect the interaction and may even enhance the ability of LDLR to remove LDL. This is because the mutations causing FH have different effects on LDLR function (classified as LDLR mutations classes 1, 2, and 5) compared with those of class 3 and 4 mutations; further research is needed to determine the effect of mutations on LDLR activity. 31 Interestingly, some interactions between APOB-100 and LDLR occur outside the normal binding sites, as shown in our docking simulations. These noncanonical interactions might reflect compensatory binding in response to mutations or could represent abnormal interactions that disrupt proper LDLR function.32,33 By altering the efficiency of LDL uptake or promoting dysfunctional cholesterol metabolism, these aberrant interactions may play a critical role in the development of FH. However, this needs to be further evaluated through molecular dynamic simulations. Overall, targeting specific interaction sites contributing to the reduced binding affinity and elevated LDL-C levels may offer potential avenues for CVD treatment.
The innovative approach employed in this study not only helped comprehend CVD but also offered valuable insights into potential therapeutic strategies. We comprehensively investigated the genetics of LDLR and APOB-100 proteins and provided previously undiscovered genetic insights. Compared with other studies in this domain, our study focused on the binding affinities and molecular interactions between these proteins. This innovative approach has the potential to revolutionize our understanding and treatment of CVDs, representing a noteworthy advancement in our efforts to provide personalized and highly effective medical therapies to individuals with heart-related conditions.
This study has some limitations. Although in silico molecular docking and computational modeling provide valuable insights, real-world complexities may vary; therefore, experimental validation is required to substantiate these findings. We conducted extensive literature review and found that the known binding sites for these proteins are scarce. Therefore, we opted for a more computational approach. We also compared multiple tools but did not report it in this article. The main reason to use GRaSP is that it can deal with bigger structures, which most tools may not be able to manage. This study focused on specific genetic variants unique to the Vietnamese population with FH, which could potentially limit the generalizability of the results to broader populations. Furthermore, we primarily examined binding affinities and interactions, highlighting the need for further research into the downstream cellular and physiological effects. These considerations highlight the importance of continued investigation and validation in both experimental settings and diverse populations with FH. Despite these limitations, this study represents a remarkable advancement in our ongoing endeavor to elucidate the complexities of FH and offers prospects for enhanced health care outcomes.
Conclusions
We identified novel LDLR and APOB protein mutations, disrupting LDL-C binding and contributing to FH pathogenesis. We identified previously unrecognized binding residues exclusive to the wild-type and precursor isoforms of LDLR, highlighting their role in disease progression. Moreover, some APOB-LDLR mutants exhibited significantly low binding affinity levels and hydrogen-bonding interactions, implying stronger and potentially more detrimental interactions between them. Impaired binding affinities could substantially disrupt cholesterol homeostasis, potentially impeding normal LDL internalization and cholesterol utilization, which is crucial in CVD progression, through LDL-C accumulation in the bloodstream. These interactions may represent promising therapeutic targets in managing FH. We identified crucial hydrogen bond interactions involving a mutated arginine residue of LDLR. This novel site represents a potential avenue for lipid-lowering therapy to restore the binding affinity and reduce LDL-C levels. Collectively, our findings highlight novel variations in LDLR and APOB-100 proteins and their relevance to disease onset. Despite inherent limitations, these insights expand our understanding of CVD development, aid in identifying novel targets for further investigations, and offer potential avenues for improved health care outcomes.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322241301267 – Supplemental material for Exploring LDLR-APOB Interactions in Familial Hypercholesterolemia in the Vietnamese Population: A Protein-Protein Docking Approach
Supplemental material, sj-docx-1-bbi-10.1177_11779322241301267 for Exploring LDLR-APOB Interactions in Familial Hypercholesterolemia in the Vietnamese Population: A Protein-Protein Docking Approach by Ngoc-Thanh Kim, Doan-Loi Do, Mai-Ngoc Thi Nguyen, Hong-An Le, Thanh-Tung Le and Thanh-Huong Truong in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-2-bbi-10.1177_11779322241301267 – Supplemental material for Exploring LDLR-APOB Interactions in Familial Hypercholesterolemia in the Vietnamese Population: A Protein-Protein Docking Approach
Supplemental material, sj-xlsx-2-bbi-10.1177_11779322241301267 for Exploring LDLR-APOB Interactions in Familial Hypercholesterolemia in the Vietnamese Population: A Protein-Protein Docking Approach by Ngoc-Thanh Kim, Doan-Loi Do, Mai-Ngoc Thi Nguyen, Hong-An Le, Thanh-Tung Le and Thanh-Huong Truong in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
The authors acknowledge the colleagues from the “10 Countries Study” and the EAS-FHSC for their support to establish the VINAFH Registry. The authors thank the Department of Clinical Biochemistry, PathWest Laboratory Medicine (Western Australia), Department of Medical Biology and Genetics, Hanoi Medical University, BIMEDTECH Molecular Biology Laboratory, and Gene Solutions (Ho Chi Minh City) for genetic testing. The authors also thank BioCode for consultation and technical support regarding protein models and molecular dynamic simulations. This study was funded by the VINAFH program for genetic testing.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the VINAFH program for genetic testing.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
T-HT and N-TK initiated the study, designed data collection tools, monitored data collection, cleaned and analyzed the data, and drafted and revised the manuscript. D-LD, M-NTN, H-AL, and T-TL monitored data collection and revised the manuscript. All authors read and approved the final manuscript.
Ethical Approval
This study was conducted according to the guidelines of the Declaration of Helsinki and was approved by the Ethics Committee of Hanoi Medical University (approval number: 39/GCN-HĐĐĐNCYSH-ĐHYHN, approval date: April 19, 2021).
Consent to Participate
Informed consent for using genetic data was obtained from all patients with FH or from the parents or legal representatives for those aged <18 years.
Data Availability
Further inquiries should be directed to the corresponding authors.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.