
Abstract
CD36 is an integral membrane protein which is thought to have a hairpin-like structure with alpha-helices at the C and N terminals projecting through the membrane as well as a larger extracellular loop. This receptor interacts with a number of ligands including oxidized low density lipoprotein and long chain fatty acids (LCFAs). It is also implicated in lipid metabolism and heart diseases. It is therefore important to determine the 3D structure of the CD36 site involved in lipid binding. In this study, we predict the 3D structure of the fatty acid (FA) binding site [127–279 aa] of the CD36 receptor based on homology modeling with X-ray structure of Human Muscle Fatty Acid Binding Protein (PDB code: 1HMT). Qualitative and quantitative analysis of the resulting model suggests that this model was reliable and stable, taking in consideration over 97.8% of the residues in the most favored regions as well as the significant overall quality factor. Protein analysis, which relied on the secondary structure prediction of the target sequence and the comparison of 1HMT and CD36 [127–279 aa] secondary structures, led to the determination of the amino acid sequence consensus. These results also led to the identification of the functional sites on CD36 and revealed the presence of residues which may play a major role during ligand-protein interactions.
Introduction
Long chain fatty acids (LCFAs) are an important source of energy for the body. Disruption of their plasma concentration is a determining factor in the onset and development of some diseases, mainly cardiovascular and metabolic ones. The membrane transport, which is the first step toward LCFA utilization consists of two components: diffusion “flip-flop” 1 and protein facilitated transfer, such as CD36. 2 The altered expression of human CD36 is directly associated with dyslipidemia and the development of some cardiovascular diseases. For example, the CD36 deficiency among Japanese population correlates with the development of hypertension, insulin resistance, and an abnormal concentration of plasma lipids.3,4 CD36 polymorphisms were also shown to be correlated with dyslipidemia and abnormal concentration of FAs in Caucasian populations. 5 In vitro, lipid incorporation was altered in muscle and adipose tissues of CD36-null mice and SHRs.6,7
CD36 is highly expressed in tissues that require energy of FA oxidation, such as the heart and skeletal muscles.8,9 CD36 is also expressed in adipose tissue 10 and in pneumocytes characterized by the absorption of palmitic acid. 11 CD36 is most abundant in proximal segments and in villi enterocytes, where most lipid absorption occurs. 12 The tissue distribution of CD36, and the research cited above, confirm the role of this receptor in uptake and membrane transport of LCFAs. However, the non-resolution of the 3D structure of CD36 remains a large hurdle in expanding studies on this protein. The first study to elucidate the CD36-FA binding site was done by Baillie et al, 13 suggesting a potential site by utilizing a simple sequence alignment and without any validation.
The present study was designed to predict the 3D structure of CD36 [127–279 aa] based on homology modeling with the FABP-H.
Methods
Sequence retrieval.
The target sequence 127–279 of CD36 was obtained from FASTA sequence of (CD36_HUMAN) Platelet glycoprotein 4 with 472 amino acids (aa) encoded by P16671 in Uniprot database. 14
Template identification and sequence alignment.
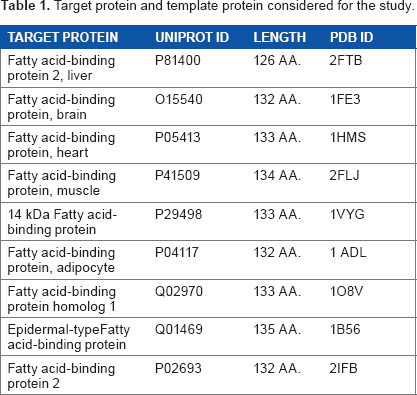
The CD36 searched for similar sequence using the NCBI BLAST 15 (Basic Local Alignment Search Tool). This search gave no significant results for the entire extracellular portion [30–440 aa] of CD36 as the binding site for FAs predicted [127–279 aa], reason why we decided to choose FABPs (2FTB, 1FE3, 1HMT, 2FLJ, 1VYG, 1ADL, 1O8V, 1B56, 2IFB) (Table 1) which have the same characteristic to bind LCFAs but showed alignment scores below 30%.
Target protein and template protein considered for the study.
Human Muscle Fatty Acid Binding Protein (H-FABP) with 133 aa (PDB code: 1HMT) binds several LCFAs, as with the case of target sequences. The functional characteristics similarity between H-FABP and CD36 [127–279 aa], the high-resolution of 1HMT with resolution value of 1.4 Å, and the results of a study on reversible binding of LCFAs to purified fat, the adipose CD36 homolog, 13 are the why 1HMT was selected as a template.
Comparative modeling and structure refinement.
The theoretical structure of CD36-FA binding site of 1HMT was generated using MODELLER-9v11 16 by comparative modeling of protein structure prediction.
MODELLER implements comparative protein structure modeling by satisfaction of spatial restraints. The program was designed to use as many different types of information about the target sequence as possible. 17 MODELLER generated several preliminary models which were ranked based on their DOPE scores. Several models with the lowest DOPE scores were selected and the stereochemical properties of each one were assessed by PROCHECK, 18 and Errat. 19 The model is visualized by Discovery Studio Software. 20
PROCHECK analysis of the model was performed to determine whether the residues were falling in the most favored region of the Ramachandran plot. 21 The model with the least number of residues in the disallowed region was selected for further studies. Errat was used for verification of evaluating the progress of crystallographic model building and refinement.
Calculation of root mean square deviation (RMSD) was performed by UCSF Chimera program. 22 It determines the best-aligning pair of chains between template and match structure.
Comparison between secondary structures of target/template.
Secondary structures of CD36 [127–279aa] and 1HMT were compared using UCSF Chimera program, which gives the consensus sequence.
Prediction of secondary structure.
PSIPRED, 23 BetaTPred2, 24 and GAMMAPRED 25 servers were used to predict the secondary structure of the target sequence, recognize helix, and strand and coil regions.
Expasy ProtParam server 26 determined the percentage of amino acid components essential for interpretation of the predicted units of secondary structure.
Functional site prediction.
Q-Sitefinder server 27 was used to predict binding site residues in modeled target sequences. Ten binding sites were predicted for the target sequence. These binding sites were further compared to the active sites of the template.
Pocket-Finder 28 was used to compare pocket detection with our ligand binding site detected by Q-sitefinder.
Results
Qualitative study of predicted model for CD36-FA binding site.
The generated model was confirmed using NIH SAVeS (Structural Analysis and Verification Server) (Fig. 1) and the accuracy was judged by PROCHECK analysis, which showed that 96.4% of the residues were found in allowed regions of the Ramachandran Plot (Fig. 2). Among the 138 residues, 128 residues were found in the most favored region, 5 in the additionally allowed region, 2 in the generously allowed region, and 3 residues in the disallowed region (Table 2).
Ramachandran plot calculations of CD36 [127–279 aa].


Modeled structure of CD36 [127–279 aa] where α-helices have been represented by red color, strands by cyan, loops in green and the gray color indicates the coil.
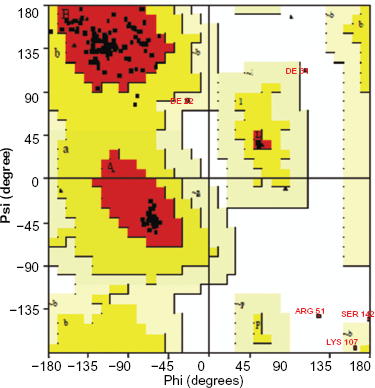
Ramachandran plot graphical.
Overall quality factor was calculated with Errat analysis and the modeled structure was found to have 53.103% quality factor. The RMSD between 102 atom pairs of predicted model and template is 0.605 Å (Fig. 3).
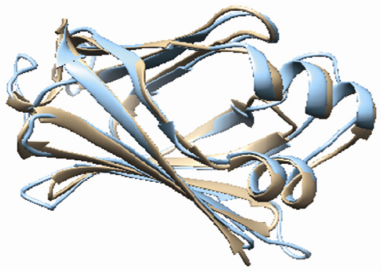
Superposed structure of Target (Tan) and Template (Cyan).
Secondary structure prediction.
Table 3 shows the data obtained from the Expasy's ProtParam server, giving the percentage of amino acid components of the target sequence. The high percentage of proline, glycine, aspartic acid, and asparagine in the CD36 [127–279 aa] demonstrates the dominance of beta turns in this molecule compared with gamma turns, which are clear in the results of BetaTPred2 and GammaPred servers (Fig. 4).

(a) Predicted beta turn residues and (b) gamma turn residues of CD36 [127–279 aa].
Percentage amino acids components of the target sequence.

PsiPred server revealed 19.6% residues in the formation of two helices, 23.52% residues in six strands, and 56.88% residues in formed coils. Comparison of match CD36 [127–279 aa] and 1HMT shows a strong similarity of their secondary structure, with the exception of a short part in the upstream, the sequence length of beta sheets, and parts of gaps (Fig. 5).

Comparison between secondary structure of target and template, structure helices in yellow, green residues present structure strands and orange for fully populated columns. Consensus presented by letters in red.
Using the UCSF Chimera program, we were able to determine 22 consensus residues between the matched CD36 [127–279 aa] and 1HMT (Fig. 5).
Determination of CD36 [127–279aa] active site.
Among the proposed sites obtained by Q-SiteFinder, and based on the comparison of CD36 [127–279 aa] with the FA binding site of 1HMT, (CD36-S1) site was identified as: ALA143, SER146, TYR149, GLN152, PHE153, LEU158, ILE162, ASN163, LYS164, LYS166, SER167, SER168, PHE170, GLN171, VAL172, THR174, ARG176, LEU189, PRO191, PRO193, THR195, THR196, THR197, VAL198, TYR212, LYS213, VAL214, PHE215, LYS218, ASP219, TYR238, GLU240, SER253, PRO255, LEU264, PHE266, SER274, TYR276 (Fig. 6).

Predicted site1, ligand colored in yellow and site residues in brown.
(CD36-S1 site) occupies a volume of 719 Å 3 with 5.16% of the total volume of target sequence.
Pocket-Finder, which uses the same interface as Q-SiteFinder, predicted that the site volume of the pocket has 1255 Å 3 with 9% of the total volume of the target sequence. This site contains the same residues of (CD36-S1 site) and the complementary residues that are crucial for the building of the pocket.
Discussion
In this study, we determined the 3D model of CD36-FA binding site. Qualitative analysis of the predicted model presented the best quality model, which was reliable as 97.8% residues were in allowed regions according to Ramachandran plot statistics results. Errat analysis showed that the predicted 3D structure is stable, in addition to the high similarity in the 3D structure determined by the RMSD of superimposed template-target.
The secondary structure prediction, using Psipred, validated the target model based on their 2D structure.
The comparison between the predicted 2D structure and that obtained by UCSF Chimera program confirm the similarity of the 2D structure category in certain portions and their location in the same position and was refer to by the helix [140–164 aa] and the three strands [225–230 aa], [262–267 aa], and [273–279 aa]. According to the functional site prediction mentioned above, we were able to determine that these six residues (Ser146, Ser168, Tr174, Leu189, Leu264, and Tyr276) are involved in the formation of the active site.
In this paper, we were interested in the (CD36-S1) site, which is probably the most active site for fixing LCFA. However, it is necessary to take into consideration the importance of other active sites predicted in the interaction between LCFA and CD36.
Recently, Kudal et al. showed that Lys164 of CD36 plays a critical role in uptake of LCFAs. 28 In our studies, and after alignment analysis of the target and template, we were able to show that Lys164 is semi-conserved (data not shown). On the other hand, the position of Arg273 in the last strand suggests a role of this amino acid in CD36. We could conclude that besides the six consensus residues mentioned above, the Arg273 and Lys164, which are semi-conserved, are probably involved in binding FAs by the CD36 protein.
In the generated model, two parts were detected. The first one is composed of alpha-helices with hydrophobic characteristics allowing a high affinity for LCFAs. This part must be considered as the main portal of the CD36 receptor to LCFAs. The second part is considered a central barrel where all the functional sites predict by Q- Sitefinder are co-localizing. This part of CD36-FA binding site contains a cavity which is characterized by the presence of a hydrophobic segment [184–204 aa] and is probably associated with the cell membrane. 29
Conclusion
This paper describes for the first time a 3D homology modeling of transmembrane protein CD36 using a cytoplasmic protein template. The 3D structure prediction of CD36-FA binding site is a first step towards understanding the role played by this receptor in lipid metabolism and development of different pathologies. The predicted functional sites and the precise localization of the essential residues in LCFAs binding will allow us to start docking analysis and confirm the precise role of these CD36 residues in FA binding.
Author Contributions
Conceived and designed the experiments: ZT, OS. Analyzed the data: ZT. Wrote the first draft of the manuscript: ZT. Contributed to the writing of the manuscript: AI, OS. Agree with manuscript results and conclusions: AM, NAA. Jointly developed the structure and arguments for the paper: AI, AK. Made critical revisions and approved final version: AK, AI. All authors reviewed and approved of the final manuscript.
DISCLOSURES AND ETHICS
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.
Footnotes
Acknowledgments
We acknowledge support from Volubilis (French-Moroccan Action Intégrée) to AI & AM. The authors are grateful to Natalia Khuri from University of California at San Francisco for technical assistance.