
Abstract
Background:
Coronary artery disease (CAD) is one of the leading causes of death worldwide. The buildup of atherosclerotic plaque, including lipids and cellular waste, characterizes this disease. Smooth muscle cells (SMCs) can migrate and proliferate to form a fibrous cap that stabilizes the atherosclerotic plaque in response to plaque buildup. However, in some severe cases, the fibrous cap is unable to prevent plaque rupture, which can lead to a thrombotic event causing a stroke or myocardial infarction. Studies have been conducted to identify genes associated with this disease. However, the influence of sex on CAD risk is poorly understood due to the complexity of the disease and the lack of women in clinical studies.
Methods:
This study is investigated with a unique collection of human aortic smooth muscle cells (huASMCs) derived from 118 male and 33 female individuals who either underwent a heart transplant or were victims of motor vehicle accidents. In this investigation, we explore differentially expressed genes between males and females related to atherosclerosis using a unique RNAseq dataset of human aortic SMCs.
Results:
Our study identified 8 genes (CHST1, DKK2, DLL4, EIF1AXP1, GALNT13, NOTCH4, SELL, SPARCL1) that exhibit sex-biased effects in SMCs. Of these, 6 genes were found in the Athero-Express dataset and 5 of them were associated with atherosclerosis-relevant phenotypes. We discovered a novel NOTCH4/DLL4 pathway that plays a role in the differential expression of these genes between males and females. This pathway is linked to coronary artery physiology and may play a role in the pathophysiology of coronary artery disease that differs between the sexes.
Conclusions:
Overall, this investigation shows that differentially expressed genes between males and females in human aortic SMCs exist.
Introduction
Cardiovascular diseases (CVD) account for 31% of all deaths worldwide. Coronary artery disease (CAD) constitutes 43.2% of all CVD. 1 Environmental, lifestyle, and genetic factors contribute to the likelihood of developing CAD. As a complex disease, the genetic propensity to develop CAD depends on a combination of multiple variants that cohesively increase the risk.
Coronary artery disease is caused by plaque buildup in the walls of the arteries that supply blood to the heart, also known as coronary arteries. The plaque is made up of deposits of cholesterol, fat, and waste that build up in the walls of the artery following damage to the endothelium layer. 2 The plaque buildup causes the inside of the arteries to narrow over time, which can partially or completely block the blood flow. This process is called atherosclerosis. 3 Vascular smooth muscle cells (VSMCs) play a significant role at all stages of atherosclerotic plaque development. Vascular smooth muscle cells contribute to many different plaque cell phenotypes, including extracellular-matrix-producing cells of the fibrous cap, macrophage-like VSMCs, osteoblast-like VSMCs, and foam-like VSMCs, all contributing positively and negatively to the progression of the disease.4-6
Data from the Global Use of Strategies to Open Occluded Coronary Arteries in Acute Coronary Syndromes IIb has shown that clinically, CAD presents differently in males and females. 7 Typically, females are likely to be diagnosed with CAD 7 to 10 years later than males and have a higher expression of cardiovascular risk factors. 8 This sex difference is also seen in other diseases as women with diabetes are at a greater risk for cardiovascular complications than their male counterparts. In addition, high-density lipoprotein (HDL) cholesterol is known to implicate a higher risk of developing CAD in women than in men. 8 The Genome-Wide Association Studies (GWAS) have identified more than 300 loci that are associated with CAD. Recent sex-stratified GWAS identified 10 out of the 300 loci to have sex differences; however, the molecular mechanism of these loci still needs to be investigated in the context of CAD.9-11
Our study investigated differentially expressed genes (DEGs) in males and females using human aortic smooth muscle cell RNAseq datasets. Our results revealed 8 genes that were differentially expressed. Six out of these 8 sex-biased genes were present in the Athero-Express dataset and 5 of them showed an association with at least one of the atherosclerosis-relevant phenotypes. In addition, we identified a novel NOTCH4/DLL4 pathway involved in the differential expression of the genes in males and females. Our findings could pinpoint new therapies specific to male and female CAD treatment plans.
Materials and Methods
Cell culture
This study is investigated with a unique collection of human aortic smooth muscle cells (huASMCs) derived from 118 male and 33 female donors who either underwent a heart transplant or were victims of motor vehicle accidents. Briefly, the cells were maintained in Smooth Muscle Cell Basal Medium (SmBM, CC-3181, Lonza) supplemented with Smooth Muscle Medium-2 SingleQuots Kit (SmGM-2, CC-4149, Lonza) (complete media). By using the 1000 Genomes reference, samples were clustered by donor phenotype, and 6, 12, 64, and 69 of the individuals were determined to be of East Asian, African, Admixed American, and European ancestry, respectively. 12 Samples from all 151 donors were cultured in 2 different media conditions, with-FBS and without-FBS, as previously conducted.13,14 The with-FBS condition simulated a disease or atherosclerotic environment in which the huASMCs modulated their phenotype to be proliferative. Therefore, the with-FBS condition will be referred to as the proliferative condition. The without-FBS condition simulated a healthy arterial environment where huASMCs maintained their quiescent phenotype. Therefore, the without-FBS condition will be referred to as the quiescent condition. 12 The cells were molecularly characterized through RNA sequencing (RNAseq), which elucidates gene expression information. The Institutional Review Board of the University of Virginia approved this study.
RNA extraction, sequencing, and quantification
As previously described, total RNA was extracted using the RNeasy Micro Kit (Qiagen) and the RNase-free DNase Set.13,14 RNA integrity scores were measured by the Agilent TapeStation and all samples had a score greater than 9, reflecting high-quality RNA preparations. Libraries were prepared with the Illumina TruSeq Stranded mRNA Library Prep Kit and were sequenced to ~100 million read depth with 150 base pair paired-end reads by the Psomogen sequencing facility.13,14
To quantify gene expression, we trimmed the reads with low average Phred scores (<20) using TrimGalore. Reads were then mapped to the hg38 version of the human reference genome using STAR in 2-pass mode to increase the mapping efficiency and sensitivity and only retained uniquely mapped read pairs. Finally, gene expression was quantified by calculating the transcripts per million (TPM) for each gene using RNA-SeQC34 based on GENCODE v32 transcript annotations.13,14
Data separation (separate conditions vs combined condition)
To conduct downstream analysis, the data were investigated in 2 separate methods. The proliferative and quiescent conditions were first analyzed for sex differences in comparison to each other, for which we have named “Separate Conditions”. Next, the data from the conditions were combined, such that sex differences could be investigated regardless of the condition. This was named as “Combined Condition” for this investigation.
Filtering
Sex filtering
Before analyzing the RNAseq dataset using the programming language, RStudio, all donors were investigated for their expression of the XIST gene (a gene that reveals donor sex). Males in which the gene appeared to be upregulated were removed from the dataset. Females in which the gene appeared to be downregulated were also removed from the dataset.
Filtering by condition group
Following this, the data from the separate and combined conditions were filtered to remove outliers. Using RStudio, we identified genes with 80% or more raw counts lower than 5 as outliers and removed them to improve statistical accuracy. We then identified genes located on the X or Y chromosome and removed them using an RStudio package, BIOMART, due to their innate attribution as male or female-specific genes. 15
Differential expression analysis
LIMMA and DESEQ2 are two out of the top three R packages that are most frequently used to analyze data of differentially expressed genes. For example, a linear model and normalized RNA-seq count data are used and required for statistics in LIMMA, while the negative binomial distribution is used in DESEQ2.16-18 With different statistical methods, gene expression results can vary. The study used both methods to benefit from their advantages.
DESEQ2
The DESEQ2 software package in the RStudio was used to analyze DEGs. In the separate conditions, all 30 female donors were compared against 30 random male donors out of the 114 male donors available. This process is referred to as random partitioning. This analysis occurred 100 times in order for each male donor to be evaluated at least once. From the 100 runs conducted, the DEGs that appeared 50 or more times were included in the downstream analysis.
LIMMA
A second method used to analyze DEGs was Linear Models for Microarray and RNA-Seq Data (LIMMA), a software package in the R programming language. The package uses linear modeling and empirical Bayes moderation to assess the differential expression and perform gene set testing. 19 Similar to DESEQ2, for the separate conditions, we used random partitioning. All 30 female donors and 30 random male donors were analyzed 100 times. The DEGs identified in all the runs were then compared to one another. The DEGs accrued in at least 50 of the 100 runs were selected for downstream analysis.
Binding analysis for regulation of transcription analysis
Binding Analysis for Regulation of Transcription (BART) is a bioinformatics tool used to predict functional factors that bind at cis-regulatory regions to regulate gene expression. This tool interprets DEGs using a ChIP-seq dataset as the input. Binding Analysis for Regulation of Transcription was sourced from the website (www.bartweb.org), and the results were plotted using RStudio. 20
Athero-express carotid endarterectomy biobank
Patients who underwent carotid artery endarterectomy in 2 Dutch tertiary referral centers between 2002 and 2020 were included in this study. Study procedures included a baseline blood withdrawal, an extensive questionnaire filled in by the participants, which was then verified against medical records, and collection of carotid arterial plaque material during surgery. All patients provided written informed consent before surgery. The study was approved by the Local Medical Ethical Committee and conducted according to the Declaration of Helsinki. 21 The Athero-Express study protocol has previously been described.22-24 All available plaque bulk-RNA samples from the female donors were used, and random males in each group were selected to match the number of females in the same age groups.
Results
Gene expression analysis and quality control
We performed sex-biased DEG analysis using huSMCs derived from 151 healthy heart transplant donors (Figure 1). To ensure that the samples were correctly separated by sex, the gene XIST was used as a positive control. Females possess 2 copies of the X chromosome, while males have 1 X and 1 Y chromosome. XIST is a gene responsible for X chromosome inactivation in female cells to achieve equal dosage equilibrium with male cells and is only expressed in cells with 2 or more X chromosomes. 25 The expression of this gene was investigated among male and female donors. Donors identified to be in the wrong group were filtered out from the analysis to maintain statistical accuracy. We found that XIST is highly upregulated in females and downregulated in males, as expected; this verified the samples were correctly separated for downstream analysis (Supplementary Figure 1).
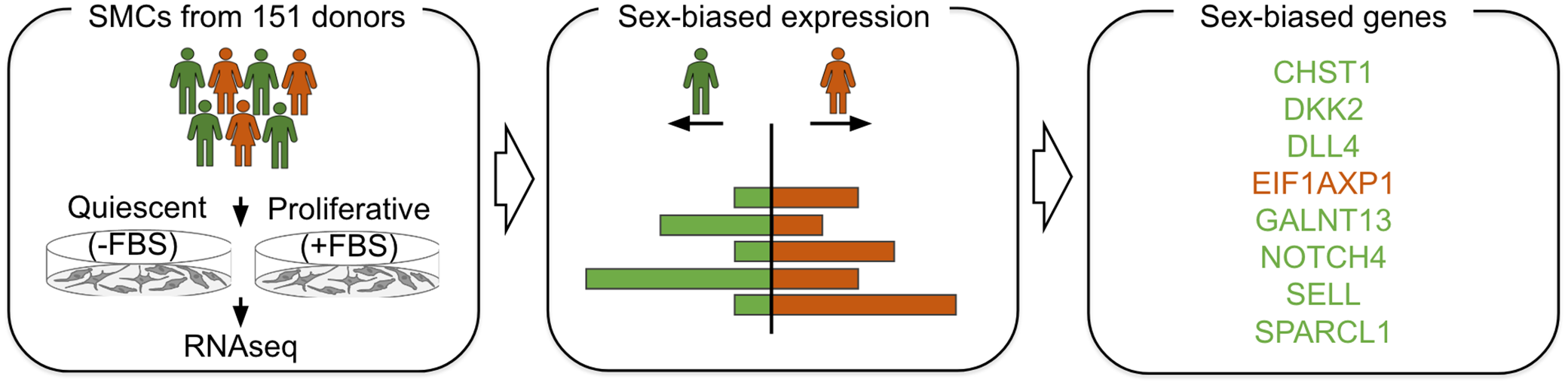
Study design and overview of analyses. We cultured human aortic smooth muscle cells (huSMCs) from 151 heart transplant donors in 2 conditions, quiescent condition (without-FBS) and proliferative condition (with-FBS). Next we conducted sex-biased gene expression analysis and identified 8 sex-biased genes. Green represents male donors and orange represents female donors.
The DESEQ2 package performs differential gene expression analysis based on a negative binomial distribution. 17 The inputs of this package include count data, which is a matrix of genes and their corresponding unnormalized counts for each sample, and a metadata file with the phenotype of the cells. 17 Our cohort of 151 huASMCl donors (142 donors post-filtering) was imbalanced due to sex. After filtering, 30 female and 112 male donors were retained. As a result of the imbalance, the analysis of the DEGs between sexes could not be performed in a typical manner. We, therefore, applied the method of random partitioning to compare the samples in a statistically and biologically significant manner. 26
Separated conditions
As a preliminary check, the imbalanced samples were graphed. There were 59 DEGs in the quiescent condition (Figure 2A) and 53 DEGs in the proliferative condition (Figure 2B) between males and females when using all 142 donors. Next, we created volcano plots to visualize the DEGs from DESEQ2, as shown in Figure 2. We considered the DEGs to be significant if they pass both the fold change cutoff of 0.5 and the P-adjusted value cutoff of 0.05. There were more downregulated genes in females, which may be attributed to the smaller number of female samples.

Volcano plots of differentially expressed genes in male and female. Plot of the genes in the quiescent (A) and proliferative (B) conditions. These were made with a P-adjusted cutoff of .05 and a fold change cutoff of .5. The gray points represent non-significant genes; genes that did not pass either the fold change cutoff or the P-value cutoff. Green points represent those genes that only passed the fold change cutoff, blue points represent those genes that only pass the P-adjusted cutoff, and red points are those that passed both cutoffs. Our results from male and female VSMCs cultured in the quiescent and the proliferative phenotype revealed more differential gene expression in the quiescent condition (59 DEGs) in comparison to the proliferative condition (53 DEGs).
Accordingly, we performed differential gene expression analysis again by applying the previously mentioned random partitioning method using 100 randomly selected cohorts consisting of 30 males and 30 females. The top 8 DEGs are shown in Table 1 with the corresponding number of positive runs out of 100. We determined genes to be significant if they were consistently differentially expressed in at least 50% of the runs (bolded). In the proliferative condition, there was 1 differentially expressed gene (NOTCH4), and in the quiescent condition, there were 4 DEGs (DLL4, SELL, CHST1, and NOTCH4). One gene, NOTCH4, was identified to be significantly differentially expressed in both conditions (Table 1). Notably, NOTCH4 and DLL4 are both differentially expressed between the 2 sexes (Figure 3A and B) and are integral parts of the same signaling pathway (Figure 3C).
Condition, Method, and Differentially Expressed Genes. Top 8 differentially expressed genes identified by LIMMA and DESEQ2 analyses. Genes with 50 or more counts (frequency) out of 100 runs are considered significant and bolded.


Differential expression of NOTCH4 and DLL4 and signaling pathways. The differential expression of the genes (A) NOTCH4 and (B) DLL4 in male and female SCM donors. (C) Schematic representation of signaling pathway involving NOTCH4 and DLL4 in the vasculature.
Combined conditions
We combined the 2 conditions (proliferative and quiescent) to identify DEGs regardless of the condition using both LIMMA and DESEQ2. Using this data of 142 donors, an example of one run is displayed as a volcano plot from DESEQ2 and can be seen in Supplementary Figure 2. In both methods, we applied the previously mentioned random partitioning method using 100 randomly selected cohorts of 30 males and 30 females. Genes were determined to be significantly differentially expressed in both conditions if they were differentially expressed at least 50 times out of 100 runs.
In addition, we considered a DEG to be significant with DESEQ2 if it passed an adjusted P-value cutoff of .05 and a fold change cutoff of .5. Similarly to DESEQ2, in LIMMA, the DEGs had to pass an adjusted P-value cutoff of .05 to be significant. The significant DEGs with both methods are displayed in Figure 4 and Table 1. With DESEQ2, we identified 3 significant DEGs: EIF1AXP1 (Figure 4A) DKK2 (Figure 4B) and SPARCL1 (Figure 4C). With LIMMA, we identified 2 significant DEGs EIF1AXP1 (Figure 4A) and GALNT13 (Figure 4D). One gene, EIF1AXP1, was identified to be significantly differentially expressed with both analysis methods.

Differentially expressed genes using LIMMA and DESEQ2 in the combined condition. With DESEQ2, we identified 3 significant DEGs: (A) EIF1AXP1, (B) DKK2, and (C) SPARCL1. With LIMMA, we identified 2 significant DEGs: (A) EIF1AXP1 and (D) GALNT13. One gene, EIF1AXP1, was identified to be significantly differentially expressed with both analysis methods.
BART
To further interpret the identified DEGs, we conducted Binding Analysis for Regulation of Transcription (BART) for the combined condition only. BART identifies transcription factors and chromatin regulators that bind at cis-regulatory regions to regulate gene expression in humans that are also potentially associated with DEGs. 20 A bubble plot of the top 15 significant transcription factors associated with the DEG is shown in Figure 5.

Results from binding analysis for regulation of transcription (BART). Transcription factors from differentially expressed genes between males and females. The top 15 significant transcription factors are shown, and they are ranked by Wilcoxon test statistic. The fold change cutoff for this method was 1. The functional factor (either a transcription factor or chromatin regulator) name is on the y-axis and the Wilcoxon test statistic is on the x-axis. The size of the dot is determined by the max area under the curve (AUC) of the gene.
Functional annotation of SMC sex-biased genes
To provide additional support for the involvement of SMC sex-biased genes in atherosclerosis-relevant phenotypes, we conducted additional lookups in the Athero-Express carotid endarterectomy biobank.24,27 From the previously identified 8 genes (CHST1, DKK2, DLL4, EIF1AXP1, GALNT13, NOTCH4, SELL, SPARCL1) that exhibit sex-biased effects in SMCs, 6 genes were also found in the Athero-Express dataset and 5 of them (CHST1, DLL4, NOTCH4, GALNT13, SELL) were associated with atherosclerosis-relevant phenotypes (Figure 6).
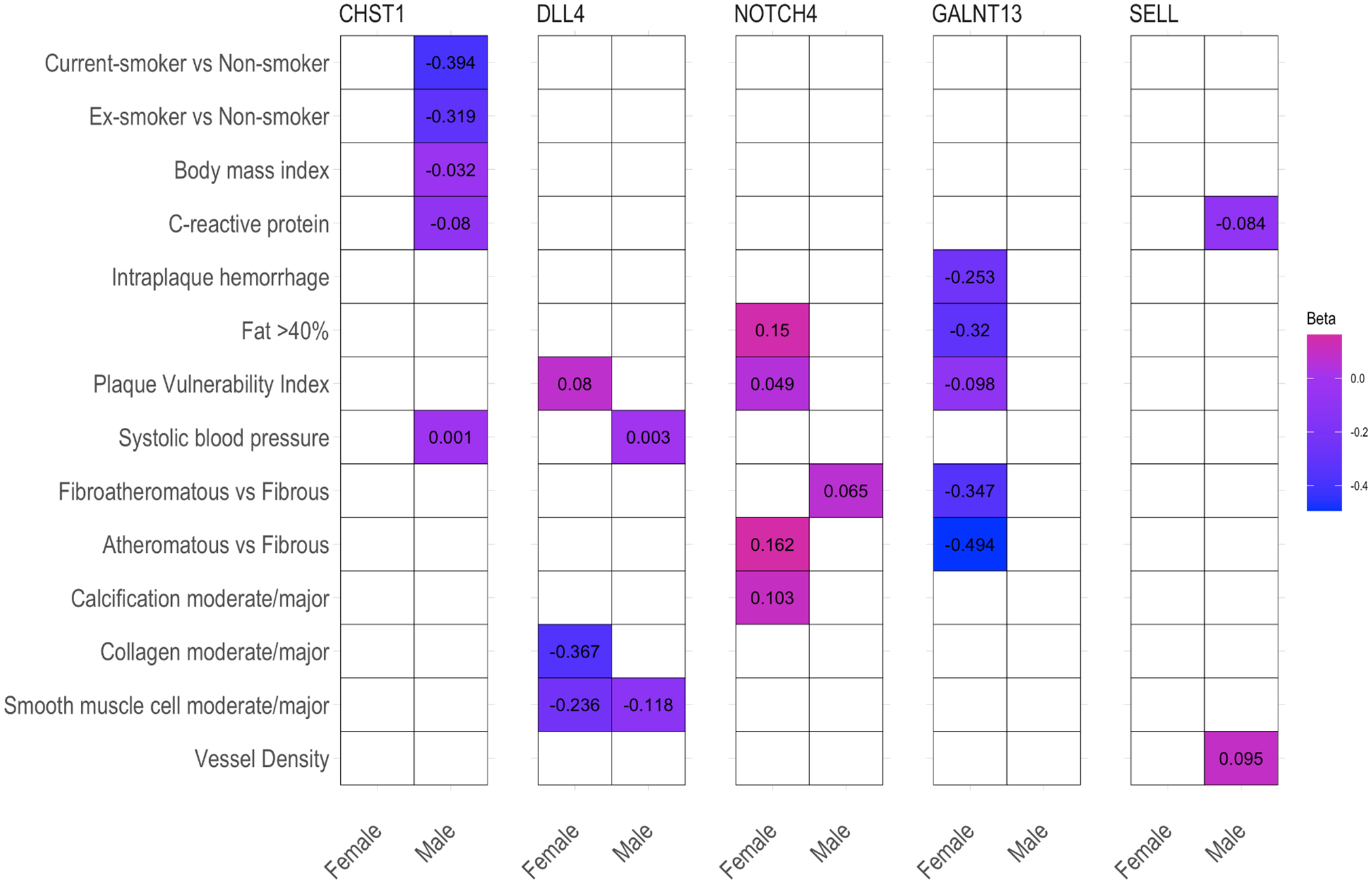
Heatmap of effect sizes (β) for significant associations between atherosclerosis-relevant phenotypes from Athero-Express carotid endarterectomy biobank and smooth muscle cell sex-biased genes. Only 6 out of the 8 significant smooth muscle cell sex-biased genes were present in the Athero-Express dataset. Five of them showed an association with at least one of the atherosclerosis-relevant phenotypes. Rows show the phenotypes and columns show the genes. The colors correspond to the effect sizes (β) ranging from −0.494 (blue) to 0.162 (pink) and only significant associations (P < .05) were shown. BMI: body mass index, CRP: C-reactive protein, IPH: intraplaque hemorrhage, PVI: plaque vulnerability index, SBP: systolic blood pressure, SMC: smooth muscle cell.
Discussion
Separate condition
DESEQ2
Overall, there were more DEGs in the quiescent condition and most of the downregulated genes were in females. However, the large number of downregulated genes in females could have been a result of the small number of female samples. Therefore, we used the method of random partitioning to remove the power imbalance between the samples. In the smaller cohorts, there were 4 genes in the quiescent condition and 1 gene in the proliferative condition that was consistently differentially expressed across the trials. There were fewer genes that were differentially expressed between males and females in the smaller cohorts. Therefore, it was determined that running the analyses on smaller cohorts was much more representative of the real DEGs than the DEGs identified from using all the donors as the power imbalance was eliminated. That was the reasoning for using this method for subsequent differential gene analyses.
In the proliferative condition, both DLL4 and NOTCH4 were differentially expressed (Figure 3A and B). These genes have been previously shown to play a role in the same signaling network (Figure 3C) Both genes were upregulated in males. We hypothesized that the pathway was differentially activated in males and females.
DLL4 plays a large role in vascular development, and it is the only NOTCH ligand that is expressed predominantly by the vascular endothelium. 28 The JAG1 gene, which is another player in this signaling network, antagonizes DLL4 and NOTCH signaling and acts downstream to promote smooth muscle cell differentiation. 28 Finally, DLL4 inactivation during coronary arterial remodeling results in small arteries, consistent with the fact that NOTCH4 promotes vascular remodeling. 28 Souilhol et al 29 used Single-cell RNA sequencing and other approaches to demonstrate that JAG1-NOTCH4 signaling promotes atherosclerosis by repressing endothelial cell subsets critical for proliferation. From previous studies linking this pathway to a role in coronary artery physiology, we hypothesized that this pathway may play a role in the pathophysiology of coronary artery disease that is different between males and females. Ultimately, while the NOTCH4/DLL4 pathway plays a significant role in the development and progression of CAD by regulating endothelial function, vascular remodeling, and plaque stability, its role is not much explored in SMC biology or sex differences.
Combined condition
DESEQ and LIMMA
In the combined condition, where we aimed to identify DEGs regardless of the condition, there are more differentially expressed genes in the DESEQ2 results as compared with LIMMA, which used a much more stringent/specific approach to identify DEGs. With both analysis methods, Eukaryotic Translation Initiation Factor 1A, X-Linked Pseudogene 1 (EIF1AXP1) was shown to be significantly differentially expressed. This gene, which is expressed in aortic tissue, has previously been shown to have sex differences in the aortic tissue through the Genotype-Tissue Expression (GTEx) project (a public resource used to study tissue-specific gene expression). 30 The findings from GTEx are consistent with our results. Unfortunately, as the gene is a pseudogene, we could not use it for downstream analysis. 31
Another significant DEG was Dickkopf WNT Signaling Pathway Inhibitor 2 (DKK2). DKK2 uses Wingless-related integration site (Wnt) signaling and is involved in embryonic development and breast cancer development. 32
In addition, we identified the transcription factor Polypeptide N-Acetylgalactosaminyltransferase13 (GALNT13). 33 Diseases associated with GALNT13 include Spheno-Orbital Meningioma and Tricuspid Valve Insufficiency. 33 It has, however, been found to function in the smooth muscle cells of the pancreas. 34 This transcription factor should be further investigated for its expression in smooth muscle cells as related to atherosclerosis.
Finally, SPARC Like 1 (SPARCL1) plays a role in the differentiation of pulmonary artery SMCs through its activation of the BMP signaling pathway. 35 Recent studies have interestingly shown that this transcription factor has heterogeneous expression in cell adhesion and/or cell migration in VSMCs located in the athero-prone aortic arch. 36 Other studies have hypothesized that SPARCL1 is an extracellular matrix protein that may have anti-adhesive and antiproliferative properties in atherosclerotic vessel walls. 37 This transcription factor should also be further investigated for its involvement in the pathogenesis of atherosclerosis in males and females.
BART
The 2 most highly ranked transcription factors we identified from the DEGs in the combined condition are both known to be sex-specific. These were Androgen Receptor (AR) and forkhead box A1 (FOXA1). AR is a transcription factor that is involved in testosterone secretion and regulates the development and growth of the prostate. 38 Targeting AR in macrophages inhibits phosphate-induced VSMC calcification by decreasing IL-6 expression. 39 Pang et al 39 state that the exact mechanism behind this process is not yet understood and question the transcription factors’ (AR and Estrogen Receptor [ER]) involvement in different sexes, as men have more coronary calcification than women. As such, our investigation sheds more light on the involvement of AR in CAD.
FOXA1 is a transcription factor that is involved in both the modulation of AR in the prostate and ER in the mammary gland of females. 40 These results indicate that the role of AR in CAD merits further investigation.
Differentially expressed genes between males and females in huASMCs do exist, and therefore, there might be a genetic difference that leads to the risk of coronary artery disease in both sexes. However, further investigation of the genetic regulation of gene expression between males and females will need to be performed by methods like expression quantitative trait locus mapping (eQTL). Further investigation of the significance and regulation of the NOTCH4 pathway as related to sex differences in CAD needs to be completed, as the mechanisms identified in this article seem promising. In addition, the gene expression of SPARCL1 in our dataset presented evidence that it is indeed differentially expressed in males and females. Further investigation of this gene also needs to be conducted. Finally, the transcription factors AR and FOXA1 have evidence that points to a common pathway that affects atherosclerosis differently in males and females. This pathway should also be further explored.
Limitations
In both separate and combined conditions, some DEGs in SMCs are also genes typically found in endothelial cells, such as SELL, NOTCH4, and VWF. Even though these genes are still expressed in VSMCs, their presence could indicate a transition to a state that combines characteristics of stem cells and endothelial cells rather than contamination. In our previous study, we observed through pathway analysis and expression profiles that these genetic modules represent VSMCs transitioning from a dormant state to a phenotype resembling atherosclerotic behavior. 41
This study has several limitations that should be considered when interpreting the results. First, there is an uneven distribution of male and female samples in our samples. This sex imbalance may introduce bias and limit the generalizability of our findings across sexes. More female samples should be added to balance the sexes to limit bias. Second, the study was done in vitro, and the results may not fully correlate with in vivo conditions. Follow-up studies should be conducted to further validate our findings in a more physiologically relevant environment. Third, the sample size used in this study, while sufficient for initial observations, could be increased in future research to enhance statistical power. A larger sample size would allow for more robust statistical analyses and potentially uncover subtle effects that may have been missed in the current study. Finally, we acknowledge that there may be other clinical information about the samples that was not available or considered in this study. These unknown factors could potentially act as confounding variables, influencing the results in ways we cannot account for. Future studies should aim to collect and analyze a more comprehensive set of clinical data to control for these potential confounders and strengthen the validity of the findings. Addressing these limitations in future research will be crucial for advancing our understanding of the sex differences in CAD and validating the conclusions drawn from this study.
Conclusions
In this study, we investigated the transcriptomic differences between male and female huASMCs in quiescent and proliferative conditions to elucidate any sex differences in atherosclerosis. Our findings revealed significant differentially expressed genes in both conditions. In the proliferative condition, we observed an upregulation of DLL4 and NOTCH4 in males. These genes are known to be involved in vascular development and remodeling, suggesting a potential sex-specific activation of this pathway in CAD pathophysiology. Analysis of the combined condition identified several significant differentially expressed genes, including EIF1AXP1, DKK2, GALNT13, and SPARCL1. These genes have various associations with vascular biology and warrant further investigation in the context of sex differences in atherosclerosis. Transcription factor analysis using BART highlighted the importance of Androgen Receptor (AR) and FOXA1, both known to be sex-specific. The involvement of AR in VSMC calcification and its potential role in CAD should be explored further.
Future studies should investigate the role of the NOTCH4 pathway, SPARCL1, AR, and FOXA1 pathways in sex differences in CAD. In addition, using expression quantitative trait locus mapping (eQTL) can help better understand the genetic regulation of gene expression between males and females. Our findings form a basis for a more comprehensive understanding of sex-specific genetic factors in CAD, potentially leading to improved risk assessment and targeted therapies for both male and female patients. Taken together, our study provides evidence for the existence of differentially expressed genes between males and females in huASMCs, suggesting a genetic basis for sex differences in CAD risk.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322241298592 – Supplemental material for System Genetics Analysis Reveals Sex Differences in Human Aortic Smooth Muscle Gene Expression
Supplemental material, sj-docx-1-bbi-10.1177_11779322241298592 for System Genetics Analysis Reveals Sex Differences in Human Aortic Smooth Muscle Gene Expression by Sarah L Meng, Rita Anane-Wae, Ernest Diez Benavente and Redouane Aherrahrou in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by an American Heart Association Postdoctoral Fellowship 18POST33990046 (to R.A.), the University of Eastern Finland (Researcher Fellowship, to R.A.), the Finnish Foundation for Cardiovascular Research (to R.A.), German Centre for Cardiovascular Research (DZHK) (to R.A.), and Junior Investigator Award from Foundation Leducq (to R.A.).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SLM, RAW, and RA designed research; SLM and RAW performed research, SLM and RAW wrote software, SLM and RAW analyzed data; and SLM, RAW, and RA wrote the paper.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.