
Abstract
Nicotinamide phosphoribosyltransferase (NAMPT) is the rate-limiting enzyme in the NAD+ salvage pathway. Our previous study demonstrated that deletion of NAMPT gene in projection neurons using Thy1-NAMPT−/− conditional knockout (cKO) mice causes neuronal degeneration, muscle atrophy, neuromuscular junction abnormalities, paralysis and eventually death. Here we conducted a combined metabolomic and transcriptional profiling study in vivo in an attempt to further investigate the mechanism of neuronal degeneration at metabolite and mRNA levels after NAMPT deletion. Here using steady-state metabolomics, we demonstrate that deletion of NAMPT causes a significant decrease of NAD+ metabolome and bioenergetics, a buildup of metabolic intermediates upstream of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in glycolysis, and an increase of oxidative stress. RNA-seq shows that NAMPT deletion leads to the increase of mRNA levels of enzymes in NAD metabolism, in particular PARP family of NAD+ consumption enzymes, as well as glycolytic genes Glut1, Hk2 and PFBFK3 before GAPDH. GO, KEGG and GSEA analyses show the activations of apoptosis, inflammation and immune responsive pathways and the inhibition of neuronal/synaptic function in the cKO mice. The current study suggests that increased oxidative stress, apoptosis and neuroinflammation contribute to neurodegeneration and mouse death as a direct consequence of bioenergetic stress after NAMPT deletion.
Introduction
NAD+ (the oxidized form of nicotinamide adenine dinucleotide) is a co-factor required for cellular energy metabolism in glycolysis, the tricarboxylic acid (TCA) cycle and mitochondrial oxidative phosphorylation (OXPHOS) in the electron transport chain (ETC). NAD+ is also a co-substrate for many NAD+-consuming enzymes, including sirtuins (SIRTs), Poly (ADP-ribose) polymerases (PARPs) and cluster of differentiation 38 and 157 (CD38 and CD157).1,2 SIRTs, PARPs, CD38 and CD157 generate nicotinamide (NAM), which in turn serves as a precursor for the NAD+ salvage pathway. In mammalian cells, NAD+ is synthesized through three pathways namely the de novo, the Preiss-Handler and the salvage pathways using different precursors including tryptophan (Trp), nicotinic acid (NA), NAM, and nicotinamide riboside (NR)1–3 (Figure 1(a) and (b)). It has been widely accepted that the salvage pathway is the predominant NAD+ biosynthesis pathway in mammals. In this pathway, the first step is catalyzed by the rate-limiting enzyme nicotinamide phosphoribosyltransferase (NAMPT), which condenses NAM and 5-phosphoribosyl pyrophosphate (PRPP) to nicotinamide mononucleotide (NMN), and the second step is catalyzed by nicotinamide mononucleotide adenylyltransferases (NMNATs), which converts NMN to NAD+. It is established that three isoforms of NMNAT, namely NMNAT1-3, are expressed in different tissues and subcellular compartments in mammalian cells where NMNAT1 is localized in the nucleus, NMNAT2 at the Golgi-cytoplasmic interface, and NMNAT3 in the mitochondria.4,5 The contents of NADP+, a structural analogue of NAD+, are tightly associated with NAD+. The interconversion between NAD(P)+ and the reduced form NAD(P)H is critical to regulate cellular redox homeostasis.
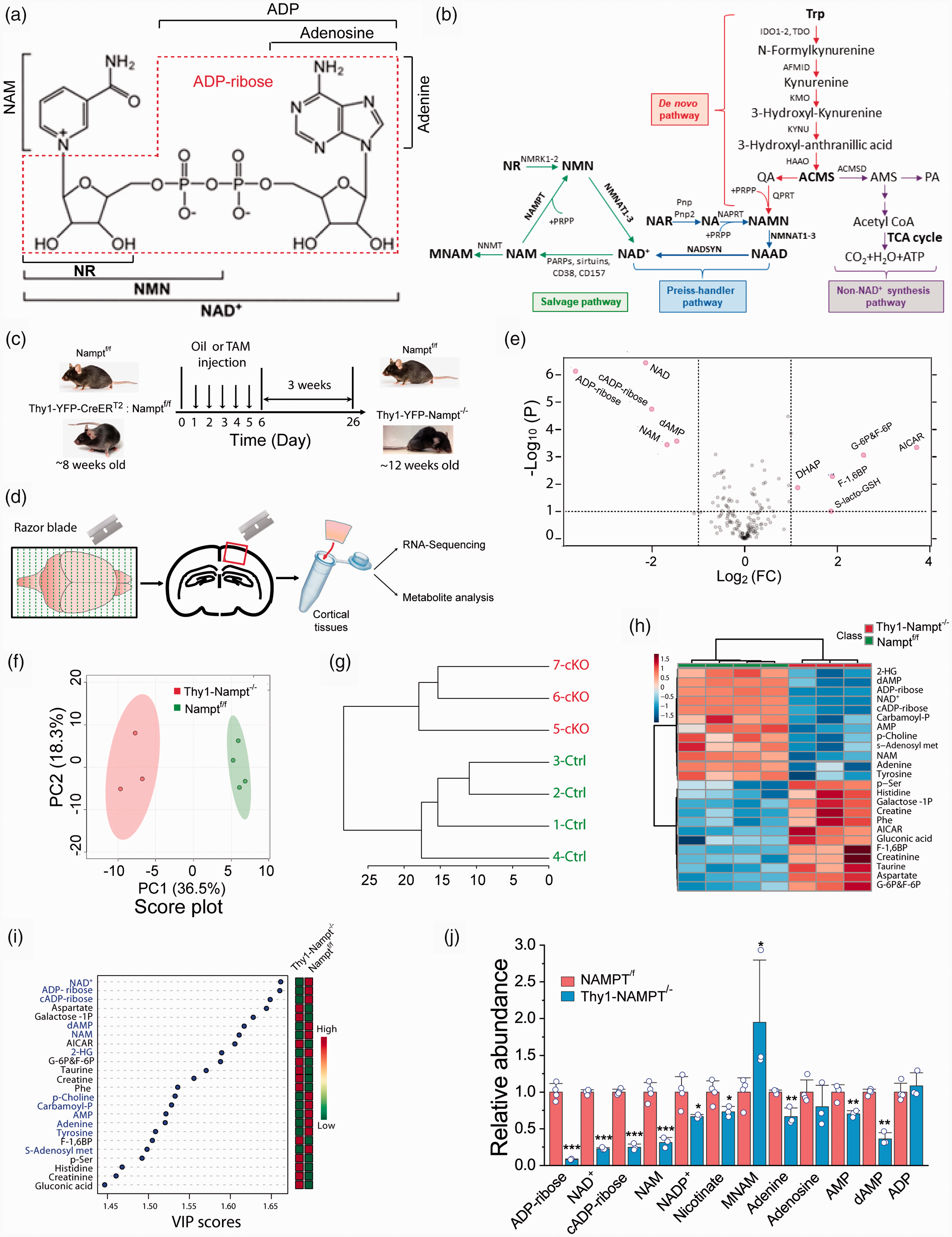
Deletion of NAMPT in the projection neurons reduces NAD+ metabolome. (a) NAD+ structure. (b) NAD+ biosynthesis pathways. (c–d) Experimental timeline. NAMPTf/f and Thy1-YFP-CreERT2:NAMPTf/f mice were injected with oil and TAM for 5 consecutive days (c) and were sacrificed for metabolomics and RNA-seq 3 weeks after the last injection (d). (e) Volcano plot of 158 metabolites with P value threshold of 0.1. FC ≥ 2 was set in volcano plot. The red circles represent features above the threshold. (f) Principal component analysis (PCA) of 158 metabolites. Top two PCs explain 54.8% (36.5% and 18.3%) of the total variance. (g) Dendrogram of metabolic tree from 158 metabolites. (h) Heatmap of top 25 metabolites. (i) Variable importance in projection (VIP) score of top 25 metabolites from partial least squares discriminant analysis (PLS-DA). The metabolites enriched in the cKO mice are labeled with blue, and reduced with black. The abundance of each metabolite is increased (red) or reduced (green) in the cKO mice is indicated in the column on the right. The colored boxes indicate the relative concentrations of the corresponding metabolites in each group. (j) Relative abundance of NAD+ metabolome. N = 4 Ctrl and 3 cKO mice. *P < 0.05, **P < 0.01, ***P < 0.005, t-test with 2-tail distribution.
NAMPT and NAD+ dysregulations have been widely reported in the non-central nervous system.2,6–8 A growing body of evidence also suggests that NAMPT plays an important role in neuronal protection in various pathological conditions in the central nervous system (CNS). Previously, we demonstrated that NAMPT is primarily expressed in neurons, but not in glial cells under normal conditions, in the mouse brain and NAMPT exerts a neuroprotective effect in ischemia through the suppression of mitochondrial dysfunction.9–11 A decrease of NAD+ in the CNS was also observed in aging and neurodegenerative diseases of human and animal models.12–14 Repletion of NAD+ precursors including NR, NAM and NMN could ameliorate pathological phenotypes of ageing related neurodegenerative diseases.15–17 Studies have demonstrated that conditional, as opposed to inducible, NAMPT deletion resulted in functional changes across a spectrum of cell and tissue types including skeletal muscle, CaMKII neurons, adipocytes, rod and cone cells.18–22 Recently, we showed that deletion of NAMPT in the projection neurons using conditional and inducible NAMPT knockout (cKO) mice, i.e., Thy1-YFP-NAMPT−/− cKO mice caused motor neuron degeneration, neuromuscular junction (NMJ) abnormalities, muscle atrophy, body weight loss, paralysis and eventually death.17,23 Our results indicate that the disruptions of bioenergetics and mitochondrial homeostasis contribute to the phenotype. To further investigate the molecular mechanisms of neuronal death in Thy1-YFP-NAMPT−/− cKO mice, we conducted a combined study of metabolomics and high-throughput RNA sequencing (RNA-Seq) to quantitatively analyze metabolic and transcriptional alterations. Using metabolomics, we studied the impact of NAMPT deletion on NAD+ metabolome, intermediate metabolites in glycolysis, the TCA cycle, pentose phosphate pathway (PPP), GSH-GSSG system and other related pathways. For the transcriptional profile study, we utilized Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of differentially expressed genes (DEGs) to perform comparative study of over-represented GO terms and KEGG enrichment pathways to elucidate the impact of NAMPT deletion. Gene set enrichment analysis (GSEA) involving all genes in the pathways was also used to analyze up and down regulated gene sets. Our study reveals the essential role of NAMPT-mediated NAD+ biosynthesis in maintaining energy and redox homeostasis in the CNS, and suggests that activation of oxidative stress, apoptosis, inflammation and immune responsive pathways are the potential mechanism of neurodegeneration after NAMPT deletion.
Materials and methods
Animals
Mice were housed in the animal facility at the University of Missouri. All experimental procedures were approved by the University of Missouri Animal Care Quality Assurance Committee, and were performed according to the NIH Guide for the Care and Use of Laboratory Animals and in compliance with the ARRIVE guidelines 2.0. 24 Both male and female mice were used in the current study. Projection neuron-specific and inducible NAMPT conditional knockout (cKO) mice were generated as described in our previous study. 17 Briefly, NAMPTf/f control (Ctrl) mice 22 were crossed with Thy1-CreERT2-YFP mice 25 to obtain NAMPTf/f:Thy1-CreERT2-YFP double homozygous bitransgenic mice confirmed by PCR amplification of their DNA from tail snips. Both NAMPTf/f and Thy1-CreERT2-YFP transgenic mouse lines have a C57BL/6J background. Deletion of NAMPT in double transgenic mice was achieved by administration of tamoxifen (TAM, 200 mg/kg in sunflower seed oil, T5648, Sigma, MO) for 5 consecutive days at 8-weeks old by oral gavage and designated as Thy1-YFP-NAMPT−/− cKO mice (hereafter, the NAMPT cKO mice). 17 Mice were sacrificed for experiments 3 weeks after the last injection of TAM.
Metabolomic and transcriptional profiling of cortical tissues
Motor cortical tissues were isolated from the Ctrl and NAMPT cKO mice for metabolomic and transcriptional profiling. Steady-state metabolomic analysis was done using LC/MS in Metabolomics Facility at UT Southwestern Medical Center (https://cri.utsw.edu/facilities/metabolomics-facility/). 26 RNA-sequencing was conducted by LC Sciences (Houston, TX). For details of procedure and analysis, see Supplemental Information.
Data analysis and statistics
Data were expressed as means ± SD. Statistical comparisons of individual metabolites and genes were made by t-test for two groups. P values were indicated in the figure legends. An assessment of normality of data were not carried out due to the nature of the in vivo study.
Results
Neuronal deletion of NAMPT reduces brain NAD+ metabolome and energy production and causes the impairment of glycolysis
In order to elucidate the molecular mechanism that potentially causes neuronal degeneration and death after NAMPT deletion in the projection neurons, 17 we conducted metabolomic study on the adult NAMPT cKO mice and the age-matched NAMPTf/f Ctrl mice to determine metabolite contents in different metabolic pathways. Three weeks after the last injection of TAM in the NAMPTf/f:Thy1-CreERT2-YFP bi-transgenic mice, motor cortical tissues were dissected for metabolomic profiling using LC/MS-based metabolomics (Figure 1(c) and (d)). Our approach allowed us to identify a total of 158 metabolites. Among them, 30 metabolites were significantly decreased and 26 metabolites significantly increased (Table S1). Univariate analysis of all metabolites using volcano plot shows up- and down-regulations of multiple metabolites in the cKO mice (Figure 1(e)). The top 5 most significantly reduced metabolites are NAD+, cADP-ribose, ADP-ribose, NAM, and dAMP which are related to NAD+ synthesis and consumption, and ATP production. The decrease in NAD+ levels in the NAMPT cKO mice is associated with ATP reduction and consistent with our previous study.10,17 Multivariate analyses using PC score, dendrogram of metabolic tree, and the heatmap and VIP scores of the top 25 metabolites show distinct phenotypic patterns between the Ctrl and cKO mice (Figure 1(f) to (i)).
Since NAMPT is the rate-limiting enzyme in the NAD+ salvage pathway, we carefully examined the contents of NAD+ and its related metabolites (hereafter, the NAD+ metabolome). Our results show that NAD+, NAM, nicotinate, NADP+, adenine, AMP, dAMP, and ADP-ribose and cADP-ribose which are the products of NAD+ generated by CD38 and CD157,27,28 were largely reduced in the cKO mice as compared with the Ctrl mice, while adenosine and ADP remained unchanged (Figure 1(j)). These data suggest that neuronal deletion of NAMPT could cause a detrimental effect on metabolic processes, redox homeostasis, energy production and signaling pathways that require NAD+ and NADP+ as a co-factor or substrate. It is important to note that methylated NAM (MNAM) is highly increased in the cKO mice (Figure 1(j)), suggesting the elimination of NAM by nicotinamide N-methyltransferase (NNMT). The increase in MNAM is also likely due to the shutdown of NAM conversation to NMN by NAMPT in the cKO mice, and consequently, the equilibrium of NAM conversion to MNAM catalyzed by NNMT shifts to MNAM side.
To further study the effect of NAMPT on neuronal bioenergetics, we conducted in vitro study using cultured neurons. Deletion of NAMPT caused reductions in NAD+, NADH and ATP levels (Figure 2(a) to (c)). Moreover, based on extracellular flux assays, both mitochondrial function and glycolytic metabolism were impaired after NAMPT deletion (Figure 2(d) to (g)). Thus, a reduction in NAD+, a direct consequence of NAMPT deletion disrupts energy production, and causes cellular bioenergetic stress and metabolic impairments.
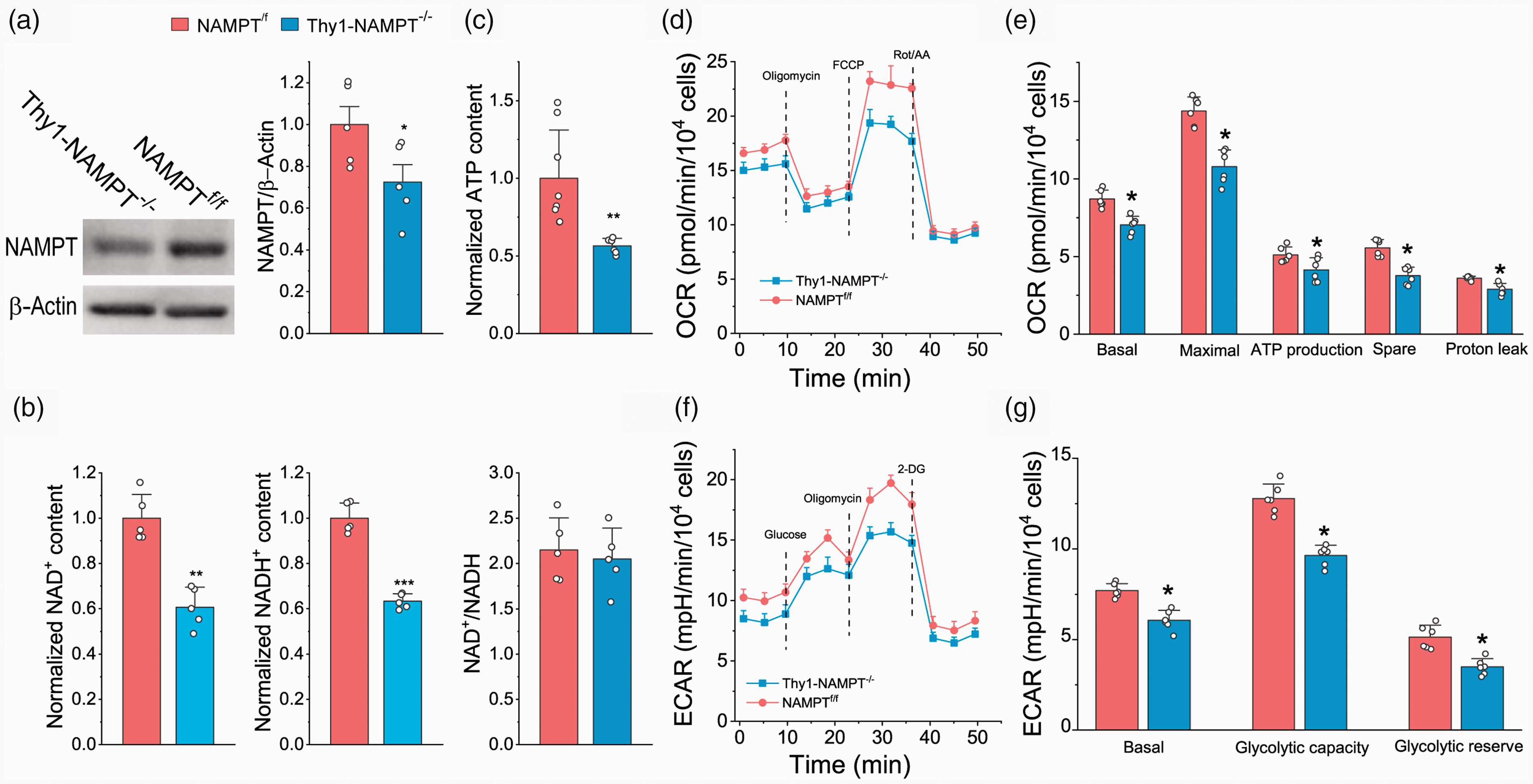
The effect of NAMPT deletion on energy production, glycolysis and mitochondrial respiration in cultured neurons. (a) Western bot image and analysis of NAMPT in cultured neurons. (b) Normalized NAD+ and NADH contents, and NAD+/NADH ratio. (c) ATP contents. (d–e) OCR traces (d) and analyses of OCR at different states (e). (f–g) ECAR traces (f) and analyses of ECAR at different states (g). Neurons were treated with 250 nM TAM for two days. Data of each group were averaged from N = 5 embryos. *P < 0.05, **P < 0.01, ***P < 0.005, t-test with 2-tail distribution.
Next, we examined the impact of NAMPT deletion on metabolites in glycolysis, the TCA cycle and other related pathways (Figure 3(a)). Our data show that the intermediate metabolites G-6P/F-6P, F-1,6BP (Fructose-1,6-biphosphate) and DHAP (dihydroxyacetone-phosphate) upstream of GAPDH in glycolysis were highly accumulated in the cKO mice, while intermediate metabolites downstream of GAPDH were not different (Figures 1(e) and 3(b)). This is likely due to the reduced enzymatic activity of NAD+-dependent GAPDH in the cKO mice. These data indicate that deletion of NAMPT causes impaired ATP production in glycolysis, further confirming cellular energy stress.
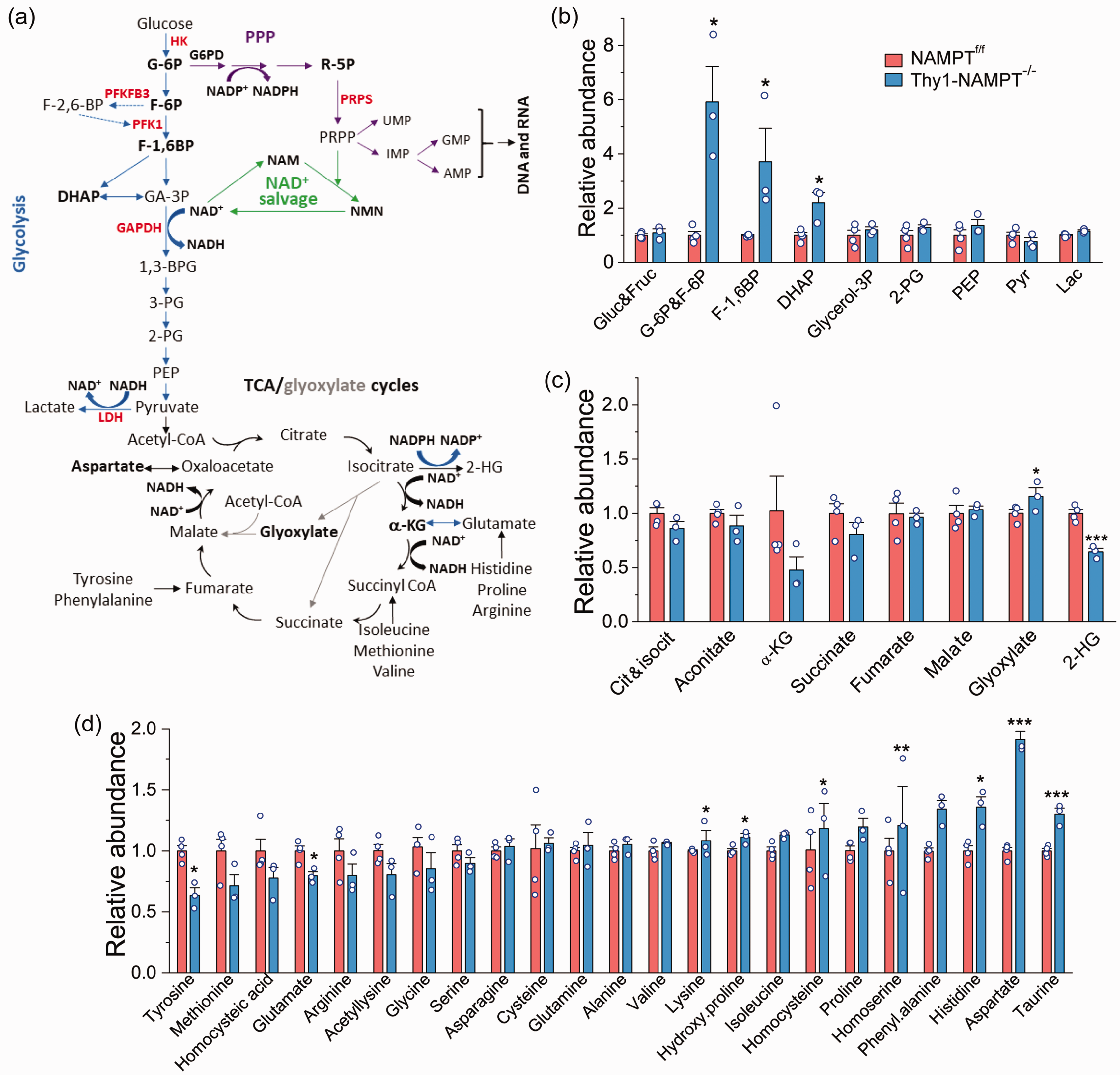
Metabolite analysis of NAMPTf/f Ctrl and Thy1-NAMPT-/- cKO mice. (a) Metabolic pathways including glycolysis, PPP, TCA/glyoxylate cycles, NAD+ salvage and AA synthesis. (b–d) Relative abundance of metabolites in glycolysis (b) and TCA/Glyoxylate cycles (c), and of AAs (d). Note: G-6P and F-6P cannot be differentiated, so the total of them were measured. N = 4 Ctrl and 3 cKO mice. *P < 0.05, **P < 0.01, ***P < 0.005, t-test with 2-tail distribution.
There was no significant change for the intermediates in the TCA cycle (Figure 3(c)). The glyoxylate cycle is a variation of the TCA cycle and an anabolic pathway, and centers on the conversion of acetyl-CoA to succinate for the synthesis of carbohydrates. 29 Here we detected that glyoxylate was significantly increased in the cKO mice. Likewise, 2-hydroxyglutarate (2-HG), which is converted from the TCA cycle intermediate isocitrate and a reductive metabolite of α-KG requiring NADPH, was also significantly reduced, presumably resulting from reduced NADPH levels.
Amino acids (AAs) are the building blocks of proteins and many of them are associated with the TCA cycle (Figure 3(a)). Among all AAs, aspartate had the highest increase in the cKO mice (Figure 3(d)). The accumulation of aspartate is likely due to the reduction of the driving step of malate to oxaloacetate (OAA) in the TCA cycle, which requires NAD+. Isoleucine, proline, phenylalanine and histidine were also significantly increased, while tyrosine, methionine and glutamate were decreased in the cKO mice. Of note, glutamate, the most important neurotransmitter and a substrate for GSH synthesis, was reduced in the cKO mice.
Deletion of NAMPT increases oxidative stress
The PPP is a metabolic pathway parallel to glycolysis and is also a major pathway for glucose metabolism (Figure 4(a)). It generates a pentose sugar, ribose 5-phosphate (R-5P), for nucleotide synthesis; another key function of the PPP is to maintain cellular redox homeostasis by producing NADPH. The glycolytic intermediate G-6P can either enter the glycolysis pathway to generate ATP or be funneled into the PPP to generate pentoses and NADPH from NADP+ in the cytosol. Noticeably, R-5P, was significantly accumulated, suggesting the buildup of G-6P causes the shuttling from glycolysis to the PPP (Figure 4(b)). R-5P can be converted to PRPP by PRPP synthetase (PRPS). PRPP is not only a precursor for the synthesis of nucleobases, nucleosides and nucleotides but is also incorporated into the NAD+ salvage pathway by NAMPT (Figures 1(b) and 4(a)). The accumulation of R-5P is also possibly due to the reduction of ATP which is involved in conversion of R-5P to PRPP. Consistent with the accumulation of R-5P, the cKO mice also exhibited reduced levels of nucleobases, nucleosides and nucleotides including adenine, AMP, dAMP, guanine, GMP, and deoxyuridine (Figures 1(e) and 4(d)).
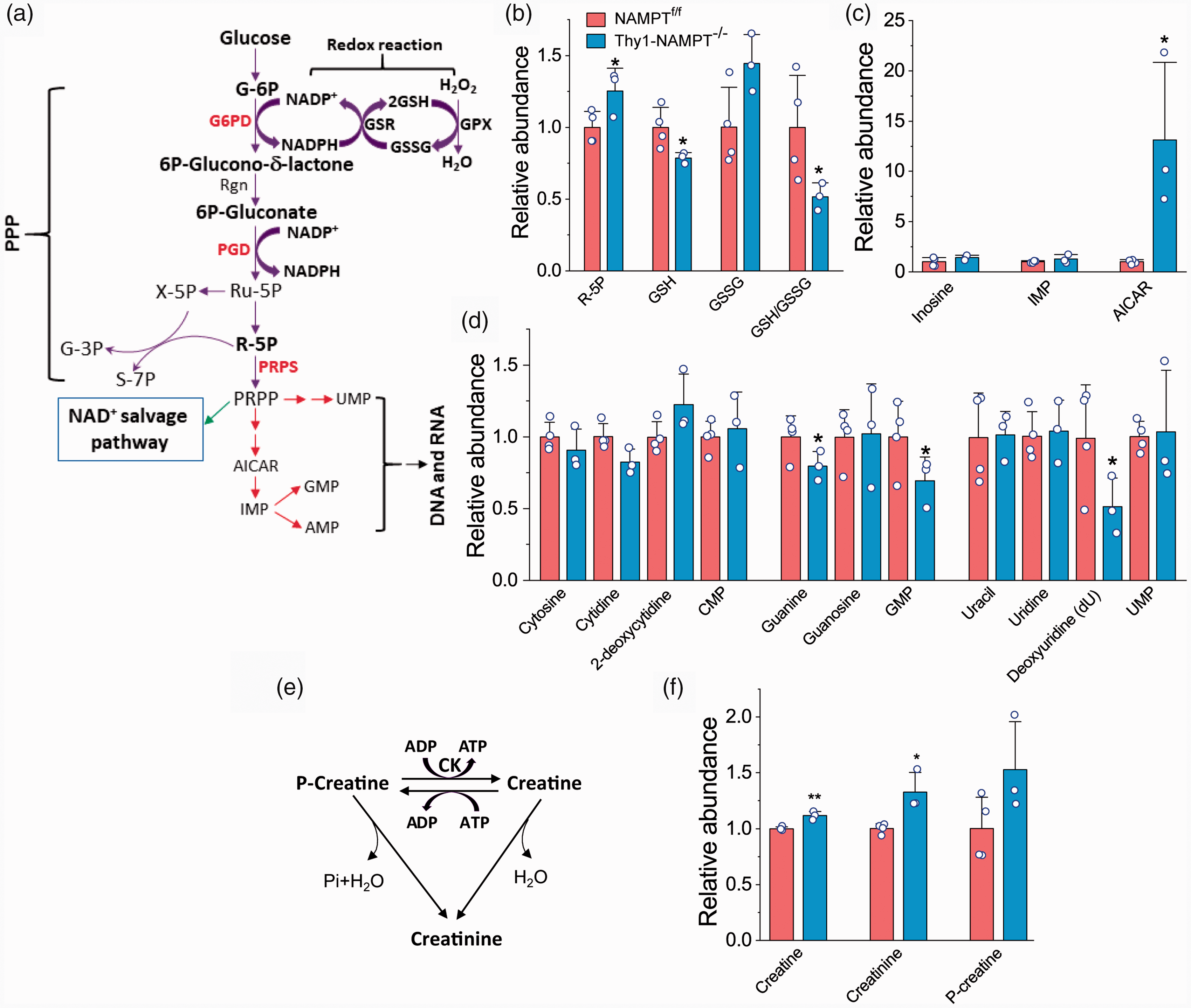
The effect of NAMPT deletion on oxidative stress, nucleotides and ATP regeneration. (a) PPP and related pathways. PPP is coupled to GSSG production through NADP. PRPP is the backbone and precursor for nucleoside and nucleotide. (b) R-5P, GSH, GSSG and GSH/GSSG ratio. (c) Inosine, inosine monophosphate (IMP) and AICAR. (d) Base, nucleoside and nucleotide. (e) Conversion among Creatine, Creatinine and P-Creatine. (f) Accumulation of Creatine, Creatinine and P-Creatine after NAMPT deletion. N = 4 Ctrl and 3 cKO mice. *P < 0.05, **P < 0.01, t-test with 2-tail distribution.
In the PPP, the conversion of G-6P to 6 P-glucono-δ-lactone is regulated by both glucose 6-phosphate dehydrogenase (G6PD) and NADP+ concentration. NADPH produced from this step is critical for maintaining cellular redox status and plays as a direct role in regulating the production of reduced glutathione (GSH), the major endogenous antioxidant produced by cells.30,31 Reduction of oxidized glutathione (GSSG) to GSH by glutathione reductase (GSR) is coupled to NADPH generated by G6PD in the PPP (Figure 4(a)). The GSH/GSSG ratio has been used as a marker of oxidative stress. Our metabolomics profiling further reveals that GSH levels and the GSH/GSSG ratio were significantly reduced in the cKO mice (21.3% and 48.7% reduction, respectively) (Figure 4(b)). The reduced GSH/GSSG ratio indicates enhanced oxidative stress in the cKO mice, probably a major reason for the neuronal degeneration and eventual death of the NAMPT cKO mice. Noticeably, S-lactoylglutathione (SLG), an intermediate related to GSH, is highly upregulated in the cKO mice (Figure 1(e)).
Meanwhile, we found that AICAR (5-aminoimidazole-4-carboxamide ribonucleotide), a downstream intermediate of PRPP, an upstream intermediate of inosine monophosphate (IMP), and an analog of adenosine monophosphate (AMP), has the highest FC increase in the cKO mice (Figures 1(e) and 4(c)). AICAR is an activator or agonist of adenosine 5′ monophosphate (AMP)-dependent protein kinase (AMPK), a key sensor of cellular energy.32,33 Given that AICAR is highly increased in the NAMPT cKO mice, it might simulate AMPK after NAMPT deletion.
ATP not only can be produced from glycolysis and OXPHOS, but also can be regenerated through the molecule phosphocreatine (P-creatine), the main storage form of energy in the cells. In this reaction, P-creatine transfers a high-energy phosphate to ADP by creatine kinase (CK), which plays a particularly important role in controlling cellular energy homeostasis (Figure 4(e)). 34 The products of this reaction are ATP and creatine, an AA. We found deletion of NAMPT caused the accumulation of creatine and creatinine (Figure 4(f)). Together with the reduction of adenine, AMP and dAMP (Figure 1(e)), our data show that deletion of NAMPT impairs ATP regeneration in the NAMPT cKO mice, causing further energy stress.
Neuronal NAMPT deletion dysregulates the expression of genes encoding enzymes in NAD metabolism
Although metabolomic analysis can estimate the contents of metabolites in various metabolic pathways, RNA-seq is considered to be one of the most effective methods for serial analyses of expression of thousands of genes encoding enzymes and proteins.35,36 Thus, we launched an RNA-seq study of motor cortex attempting to elucidate the underlying mechanism of the neuronal death caused by NAMPT deletion in the projection neurons. The RNA-seq data contained 22,611 native features. PCA shows clusters of samples to two distinct groups of the Ctrl and cKO mice (Figure 5(a)). By setting thresholds of |Log2(FC)|>1 and Padj<0.05, we found that neuronal deletion of NAMPT causes the upregulation of 1186 genes and downregulation of 418 genes with a total number of DEGs of 1604 (Table S2) as shown in Venn plot (Figure 5(b)). These data indicate that deletion of NAMPT causes a large disruption of gene expression, consistent with the distinct phenotype of the cKO mice (Wang et al, 2017).
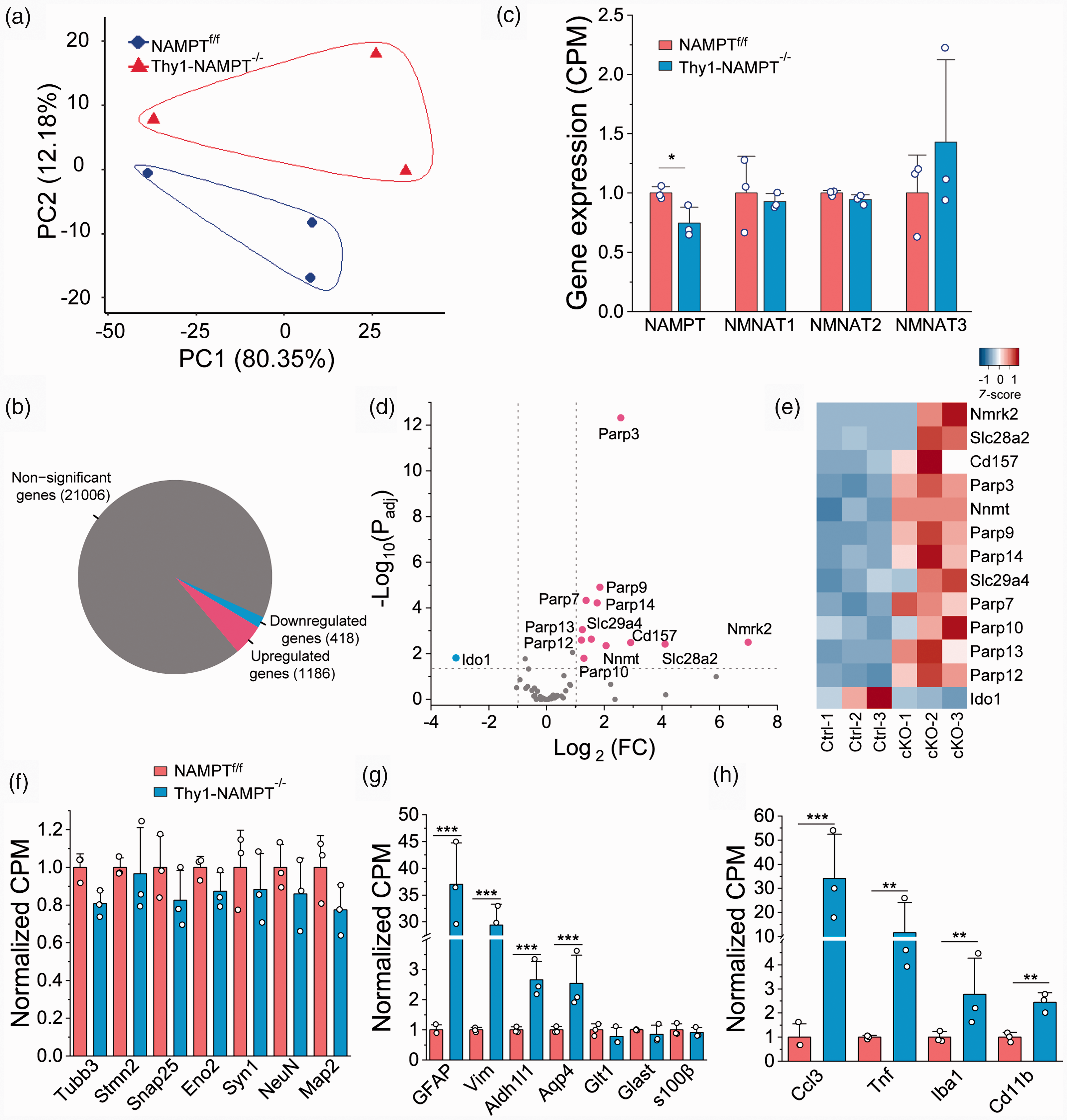
RNA-seq analysis of gene expressions involving NAD metabolism and cell-type markers in NAMPTf/f Ctrl and Thy1-NAMPT-/- cKO mice. (a) PCA analysis score plot. (b) The Venn diagram represented by differential expressed genes. (c) Relative expression of genes in the NAD+ salvage pathway. (d) Volcano plot of log2 (FC) vs log10 (Padj) showing up- and down-regulated genes involved in NAD metabolism in the cKO mice. (e) Heatmap of genes above the threshold of |Log2(FC)|>1 and Padj<0.05 in the NAD metabolism. (f–h) Relative mRNA levels of typical cellular markers of neuron (f), astrocyte (g) and microglia (h). *P < 0.05, **P < 0.005, ***P < 0.0005, t-test with 2-tail distribution. N = 3 mice in each group.
We have shown that the NAMPT cKO mice exhibited a dramatically reduced NAD+ metabolome (Figure 1(e)). To determine whether NAMPT deletion affects other genes related to NAD metabolism, we initially compared the expression of genes encoding enzymes in the salvage pathways. Indeed, the mRNA expression levels of NAMPT is reduced from 11.07 ± 0.35 in the Ctrl mice to 8.26 ± 0.85 in the cKO mice (P = 0.038, t-test), while NMNAT1-3 in the cKO mice were not significantly different from the Ctrl mice (Figure 5(c)), consistent with the reduction of NAMPT protein levels in our previous study. 17 Because NAD+ levels are determined not only by the salvage pathway but also by many other pathways including NAD+ consumption, we conducted analysis of a set of 71 genes encoding enzymes including those in NAD+ biosynthesis pathways (the salvage, de novo and Preiss-Handler pathways), NAD(H) phosphorylation, NADP(H) dephosphorylation, NAD+-dependent deacetylation, ADP-ribosylation, cADP-ribose formation, NAM methylation/oxidation, and other related functions in transport, binding, redox and regulation (Table S3). 37 The volcano plot displays up- and down-regulation of these genes (Figure 5(d)). The heatmap shows the distinct pattern of top 13 genes with |Log2(FC)|>1 and Padj<0.05 in the NAD metabolism (Figure 5(e)). Nicotinamide riboside kinases, NMRK1 and NMRK2, were considered as a part of salvage pathway and catalyze the phosphorylation of NR and nicotinic acid riboside (NaR) to form NMN and nicotinic acid mononucleotide (NaMN) (see Figure 1(b)). We found NMRK2 was significantly upregulated while NMRK1 remained similar after NAMPT deletion (Figure 5(d), Table S3). NNMT catalyzes the N-methylation of NAM and other pyridines to form pyridinium ions and its mRNA levels are largely increased (Figure 5(d) and (e)). An increased expression of NNMT will expectedly cause enhanced elimination of NAM and the increase of MNAM, thus reducing NAD+ synthesis in the salvage pathway, which is confirmed by metabolomic analysis (Figure 1(j)). In analysis of genes in de novo and Preiss-Handler pathways to determine whether there are compensatory effects for NAD+ biosynthesis in the cKO mice, only Ido1, the rate-limiting enzymes in de novo pathway was significantly reduced (Figure 5(d) and (e), Table S3). NAD+-consuming enzymes include PARPs, SIRTs and CD38/157 (Figure 1(b)). 2 Importantly, mRNA levels of several genes in the PARP family including PARP3, 7, 9-10, 12-14 were significantly upregulated but there was no significantly up- or down-regulated member of the SIRT family in the cKO mice. mRNA levels of, NAD+-consuming CD157 gene were also highly upregulated in the cKO mice (Figure 5(d) and (e)). Moreover, mRNA levels of genes Slc28a2 and Slc29a4, which encode nucleotide transporters, were highly upregulated (Figure 5(d) and (e)). Thus, NAMPT deletion causes changes in the expression of many other genes related to NAD metabolism, which may further contribute to the overall reduction of NAD+ content.
NAMPT deletion causes the upregulation of important genes in energy metabolism and redox status-related pathways
Since NAD+ is an essential co-factor in energy producing pathways, next we examined gene expression of enzymes involved in glycolysis, the TCA cycle and OXPHOS, which are critical for energy metabolism. mRNA levels of genes Glut1, Hk2 and Pfkfb3, which encode enzymes in the first 3 steps before GAPDH in glycolysis, were significantly increased in the NAMPT cKO mice (Figures 6(a) and S1(a) to (c)). Upregulations of these genes suggests that the cKO mice try coping to the energy stress by increasing glycolytic metabolism, which has a fast energy (ATP) production. Notably, the glycolytic gene GAPDH whose protein product uses NAD+ as a co-factor, remained unchanged after NAMPT deletion, and genes downstream of GAPDH were not changed either. No genes in the TCA cycle had significantly increased mRNA levels (Figure 6(b)). In OXPHOS, mRNA levels of Tcirg1, which encodes vacuolar H+-ATPase (V-ATPase) and acts as a pump to move protons across the membrane, 38 was increased; the mRNA levels of COX IV genes Cox6a2 and Cox8b, which encode the terminal enzyme of the mitochondrial respiratory chain, were reduced (Figure 6(e)). The reduced expression of COX IV genes also suggests an energy stress in the cKO mice.
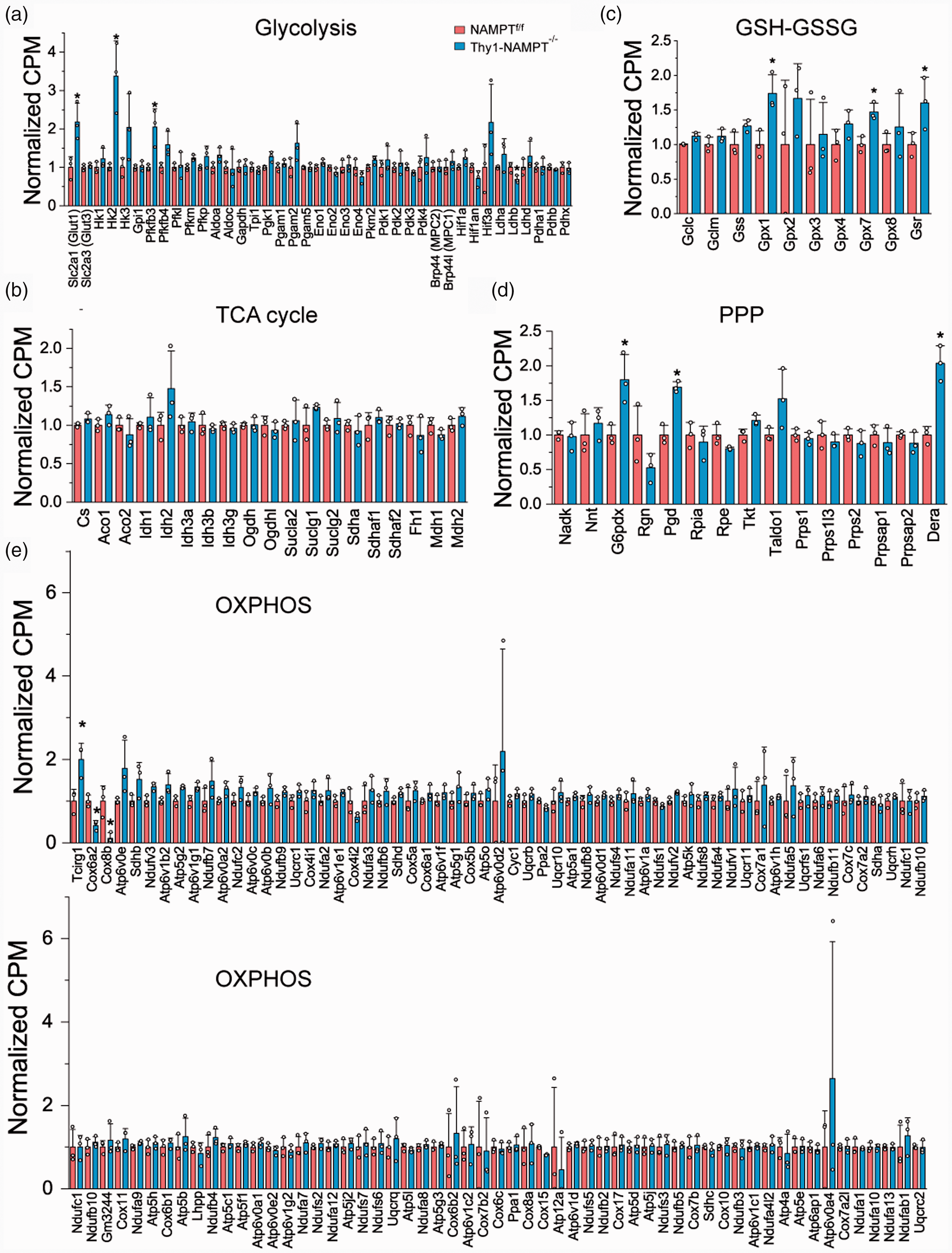
Changes of gene expression in different metabolic pathways after NAMPT deletion. (a–d) Relative mRNA levels of genes in glycolysis (a), the TCA cycle (b), GSH-GSSG system (c), PPP (d) and OXPHOS (e). *P < 0.05, t-test with 2-tail distribution. N = 3 mice in each group.
Relating to the identified metabolites of oxidative stress (Figure 4(a)), we analyzed the genes in the PPP and GSH-GSSG system that are important for maintaining redox balance and antioxidant capacity. GSR was found significantly upregulated in the cKO mice (Figures 6(c) and S1(d)), suggesting the enzymatic activity to convert oxidized glutathione GSSG to GSH, which is couple to NADPH generation by G6PD in the PPP, is increased. The proteins encoded by the glutathione peroxidase (Gpx) family catalyze the reduction of hydrogen peroxide, organic and lipid hydroperoxides, and thereby protect cells against oxidative damage. Isoforms Gpx 1 & 7 were significantly upregulated in the cKO mice (Figures 6(c) and S1(e) and (f)). G6PD (G6pdx) and phosphogluconate dehydrogenase (PDG), which encode NADP+-dependent enzymes in the PPP, were significantly increased after NAMPT deletion (Figures 5(d) and S1(g) and (h)). These results suggest that there is an attempt to increase antioxidant capacity in the cKO mice following NAMPT deletion.
Since RNA-seq was done using cortical tissues containing multiple cell types, our results only reflect an overall effect. On the other hand, tissues were collected 3 weeks after TAM administration, when the pathological conditions were fully developed, thus neuronal deletion of NAMPT likely causes gene expression changes in other cell types. To further look into this possibility, we did analysis for well-known cell-type specific genes for neurons, astrocytes and microglia (Figure 5(f) to (h)). 39 Indeed, mRNA levels of some astrocyte-specific genes including GFAP, Vimentin, Aldh1l1 and Aqp4, and microglia-specific genes Ccl3, Tnf, Iba1 and Cd11b were highly upregulated, but neuron-specific genes including Map 2, NeuN, Syn1 were not affected after NAMPT deletion.
Gene classification by gene ontology (GO) functional annotation indicates weakened immune system and impaired cellular function after NAMPT deletion
To further understand why NAMPT deletion in projection neurons causes neurodegeneration, we analyzed the functional assignment and categorization by GO enrichment analysis of DEGs using ClusterProfiler software package (http://geneontology.org/). The gene sets in GO terms of Cell Component (CC), Biological Process (BP) and Molecular Function (MF) were predicted in the significantly enriched GO terms. DEGs were involved in numerous GO terms of CC, BP and MF. Comparison between the two groups identified 58, 952 and 75 upregulated, and 48, 178 and 58 downregulated GO terms of CC, BP and MF, respectively, in the NAMPT cKO mice (Tables S4 to 6). The top 15 up- and down-regulated BP, CC and MF GO terms and their gene ratios (i.e., the number of DEGs enriched in the GO term/the number of all genes in gene sets of the input GO term from database), were displayed to reveal different distribution patterns. GO terms of CC significantly enriched in the cKO mice were a variety of extracellular matrix, cell membrane structure and subcellular compartment, and cytoskeleton and cell adhesion related components, while significantly downregulated were synapse, synaptic membrane, ion channel complex and ion transporter complex associated components (Figure S2(a) and (b), (g) to (h)). GO terms of BP significantly enriched in the cKO mice were a variety of immune- and inflammation-associated processes, while significantly downregulated were synaptic transmission, ion transport, and cognition/learning associated processes (Figure S2(c) to (d), (i) and (j)). GO terms of MF significantly increased in the cKO mice were a variety of protein binding activities (cytokine, chemokine, growth factors, receptors, integrin/fibronectin, cell adhesion molecule and extracellular matrix) associated function, while significantly decreased were ion channel and ion transporter activities associated function (Figure S2(e) and (f), (k) and (l)). Overall, GO analyses suggest a weakened immune system and impaired cellular function after NAMPT deletion in the projection neurons.
KEGG pathway analysis reveals downregulation of cellular functional pathways and upregulation of cell death and inflammation pathways
We also performed KEGG pathway enrichment analysis of DEGs using the ClusterProfiler package and KEGG pathway database (http://www.genome.jp/kegg/pathway.html). By comparing the two groups, our analysis identified 78 upregulated and 18 downregulated enriched pathways in the NAMPT cKO mice (Table S7). We found that pathways related to cell death and inflammatory immune responses including apoptosis, JAK-STAT signaling, NF-κB signaling, cellular senescence, and chemokine signaling pathways were significantly upregulated, while pathways related to neuronal and synaptic function, including neuroactive ligand receptor interaction, glutamatergic synapse, cholinergic synapse, GABAergic synapse, Ca2+ signaling and axon guidance were significantly downregulated. The top 15 up- and down-regulated KEGG pathways and their gene ratios for the cKO mice were displayed (Figure S3(a) and (b)).
GSEA shows increased activation of inflammation, immune response and cell death pathways, and reduced neuronal function after NAMPT deletion
Using a pre-ranked gene list, GSEA can determine whether an a priori defined gene set shows statistically significant and concordant differences between two phenotypes. Different from GO and KEGG pathway analyses of DEGs which are over-represented, in GSEA analysis, all the genes are provided, ranked and weighed by FC in the analysis without a cutoff. This analysis also enables the detection of subtle effects of multiple genes in the same gene set that might be missed by assessing gene individually. We conducted GSEA of 4603 metabolic pathways from the GSKB database, including REACTOME, KEGG, EHMN and MouseCye.40–43 Using our pre-ranked gene list and thresholds of NES > 1.5 and FDR q-val < 0.05, we identified 9 downregulated pathways and 206 upregulated pathways in the cKO mice (Table S8). NES of the top 5 up- and down-regulated pathways were plotted in Figure S4. Importantly, we found that apoptosis, NF-κB, cytokine-cytokine receptor interaction, JAK-STAT, and complement and-coagulation cascade signaling pathways were highly upregulated, while glutamatergic synapse, GABAergic synapse and cholinergic synapse were down-regulated in the cKO mice (Figure 7(a) and (b)). GSEA and heatmap plots of gene sets show many genes in upregulated pathways were upregulated, while many genes in downregulated pathways were downregulated (Figures 7(c) and (d) and S5). The KEGG pathways of energy production and redox-related pathways including glycolysis and gluconeogenesis, the TCA cycle, OXPHOS, PPP and glutathione metabolism were not significantly upregulated (NES < 1.5 and FDR q-val > 0.05) (Figure S6). These results demonstrated that pathways related to immune response, inflammation and cell death were upregulated, while pathways related to cellular function were downregulated after NAMPT deletion, which are consistent with GO term and KEGG pathway analyses of DEGs, and indicate that NAMPT deletion causes the inhibition of synaptic function and promotion of neurodegeneration via apoptosis and inflammation in the cKO mice.
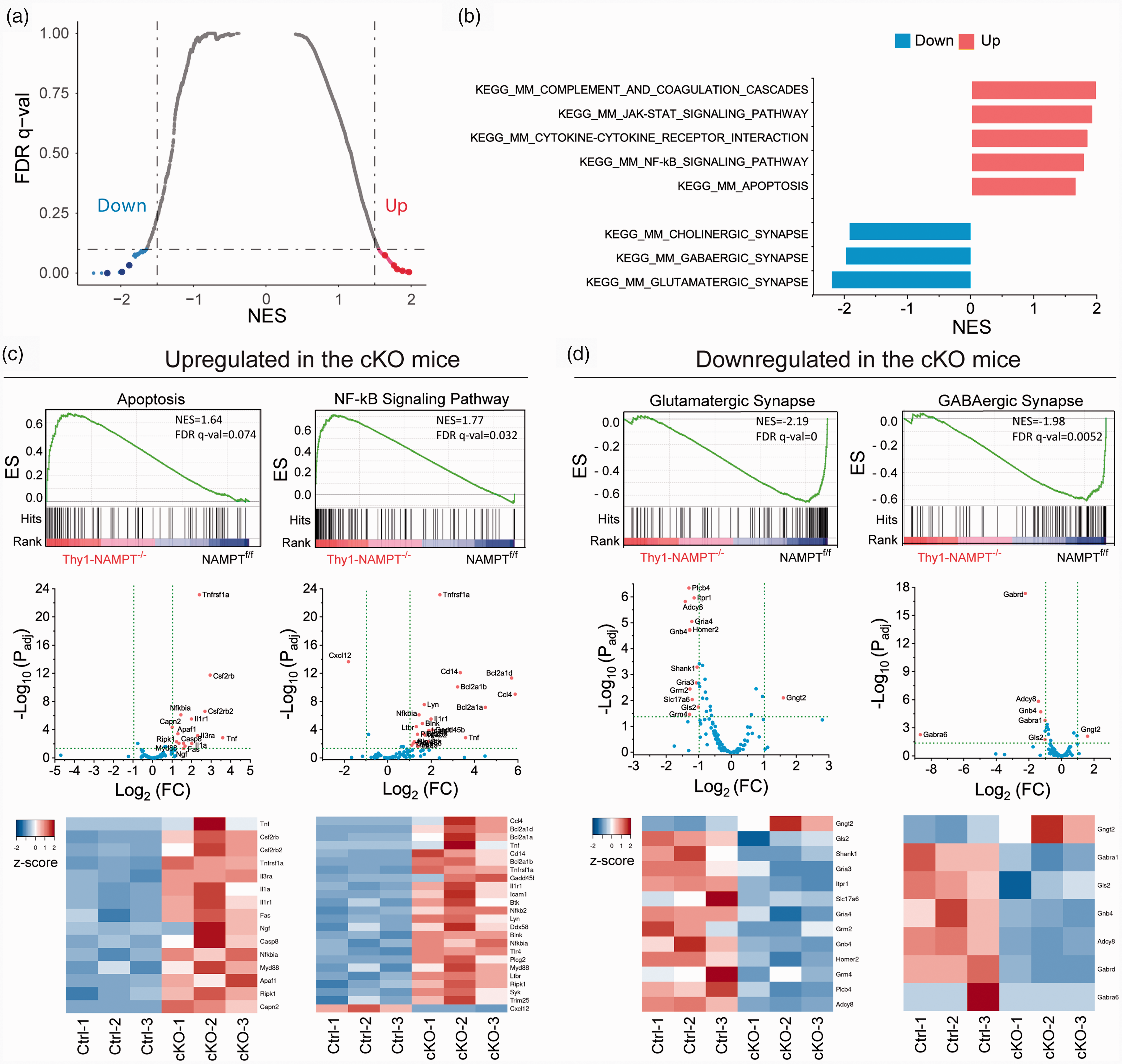
GSEA of metabolic pathways. (a) Volcano plot of false discovery rate (FDR q-value) versus NES based on GSEA of RNA-seq data from NAMPTf/f Ctrl mice and Thy1-NAMPT-/- cKO mice using 4603 gene sets of GSKB metabolic pathway gene set collection (Lai et al., 2016). Significantly down-enriched gene sets are in blue (sig=True), significantly up-enriched gene sets are in red (sig=True), and nonsignificant gene sets are in grey (sig=False). Black vertical lines highlight NES of -1.5 and 1.5, and black horizontal line represents a FDR q-value of 0.05. (b) Bar graph of FDR q-val showing representative top up-regulated (red) and down-regulated (blue) gene sets related to immune response, inflammation, cell death, and synaptic function in the cKO mice. Those pathways are visualized as large dots in A. (c–d) Representative GSEA (up), volcano (middle) and heatmap (low) plots of upregulated (c) and downregulated (d) gene sets in the cKO mice. Black bars in GSEA indicate the hits in gene sets represented among all genes pre-ranked by ranking metrics. The green dash lines in volcano plots indicate thresholds of Padj<0.05 and |Log2(FC)|>1. Heatmap plots only include genes within thresholds. N = 3 mice in each group.
Discussion
In our previous study, we demonstrated that neuronal NAMPT is an essential gene for survival using NAMPT cKO mice. 17 To further elucidate the mechanism of neurodegeneration and death of the NAMPT cKO mice, here we conducted a combined study of metabolomic and transcriptional profiling. Through comprehensive assessments of the effect of NAMPT deletion on metabolism in the motor cortex, we have obtained several novel findings. First, the NAD+ metabolome and energy production were largely reduced in the cKO mice, confirming the predominant role of NAMPT-mediated NAD+ salvage pathway in NAD+ biosynthesis and energy homeostasis. Second, G-5P/F-5P, F-1,6BP and DHAP, the metabolites before the NAD+-dependent GAPDH in glycolysis, are highly accumulated in the cKO mice, indicating that deletion of NAMPT impairs glycolytic metabolism and glycolysis is coupled to NAMPT-mediated NAD+ salvage pathway. Third, GSH levels and GSH/GSSG ratio were significantly reduced in the cKO mice, suggesting oxidative stress plays a role in neuronal degeneration. Fourth, RNA-seq study indicates that NAD+ consumption and elimination are more active in the cKO mice. Fifth, glycolytic genes Glut1, Hk2 and Pfkfb3 before GAPDH, as well as genes involved in oxidative stress, are highly upregulated in the cKO mice, corroborating the results from the metabolomic study. Sixth, GO, KEGG and targeted GSEA show that cellular function-related pathways (such as synaptic function, ion channel activity and ion transporter activity) were largely down-regulated while immune response, inflammation and cell death pathways (e.g., apoptosis, JAK-STAT, NF-κB signaling, cytokine-cytokine receptor interaction) were largely upregulated in the cKO mice, indicating their contribution to neuronal degeneration. These results provide mechanistic insights into neurodegeneration following NAMPT deletion in the projection neurons.
How does NAMPT deletion cause neuronal degeneration in the NAMPT cKO mice? A few findings indicate energy stress might be the initial cause. First, NAD+ is dramatically reduced in the cKO mice, which causes decrease in ATP production through glycolysis and OXPHOS.9,10,17 The increase in the expression of genes in NAD+ consumption pathways provides further evidence for the NAD+ reduction in the cKO mice. Second, glycolytic intermediates G-6P/F-6P, F-1,6BP and DHAP were highly accumulated in the cKO mice, suggesting the decoupling of glycolysis from NAMPT-mediated NAD+ salvage pathway and impairment of ATP production in glycolytic pathway. Third, adenine and AMP, the precursors of ATP, were reduced in the cKO mice. Fourth, the accumulation of creatine and creatinine suggests the reduction of ATP regeneration. Fifth, AICAR has the highest FC increase in the cKO mice. This is important considering AICAR is a downstream intermediate of PRPP and a stimulator of AMPK, the master regulator of energy metabolism and cellular energy stress sensor.32,33
While both genes and metabolites involved in glycolysis are significantly affected after NAMPT deletion, the analysis of the TCA cycle did not show any significant changes in either metabolites or gene expression. However, key ATP precursors and energy production are reduced in cKO mice, suggesting OXPHOS does not function normally.
Oxidative stress is associated with NAD+ depletion, a decline in metabolic regulation and reduced cell viability.11,44,45 One key function of the PPP is to maintain cellular redox homeostasis by producing NADPH, the reduced form of NADP+.31,46 NADP+ is synthesized by NAD+ kinase (NADK) by transferring a phosphate group from ATP to the 2′-hydroxyl group of the adenosine ribose moiety of NAD+ 3 and is involved in maintaining redox balance and supporting the biosynthesis of fatty acids and nucleic acids. On the other hand, reduced glutathione, GSH, a ubiquitous tripeptide with a free sulfhydryl group made up of L-cysteine, glycine, and L-glutamate, is the major endogenous intracellular and extracellular antioxidant produced by cells, participates directly in the neutralization of free radicals and reactive oxygen compounds to combat oxidative stress and maintain the normal reduced state in the cell, and is considered to be one of the most important scavengers of reactive oxygen species (ROS) to reduce oxidative stress. 47 Here, we found that GSH levels and GSH/GSSG ratio were highly reduced in the cKO mice. The GSH/GSSG ratio is an important indicator of cellular health, with a higher ratio signifying less oxidative stress in the organism and a lower ratio is indicative of neurodegenerative diseases. 3 GSH is associated with the PPP, where both G6PD and PGD, the NADP+-dependent enzymes in the oxidative phase of the PPP, play a key role in producing NADPH and converting GSSG to GSH. 48 Although the mRNA levels of G6PD and PGD are significantly increased in the cKO mice, NADP+ contents are largely reduced in the cKO mice. It is conceivable that the conversion of GSSG to GSH will be limited due to the reduction of NAPD+ levels in the cKO mice. Thus, our study indicates that NAMPT deletion could cause enhanced energy and oxidative stress, which subsequently triggers neuronal degeneration.
In light of the reduction of the NAD+ metabolome in the NAMPT cKO mice, using RNA-seq, we initially analyzed a comprehensive set of genes involving various NAD metabolism pathways including NAD+ biosynthesis, consumption and transportation. Our data show that mRNA levels of many PARP members (PARP3, 7, 9-10, 12-14) were increased, which could deplete more NAD+ in neurons and exacerbate energy stress. Similarly, the mRNA level of another NAD+-consuming enzyme BST 1 (also called CD157) is highly upregulated, which may further contribute to the reduction of NAD+ levels. We also found key glycolytic genes Glut1, Hk2 and Pfkfb3, were significantly upregulated in the cKO mice. Enhanced expression of glucose transporters and glycolytic enzymes is a common feature of metabolic adaptation to glycolysis. 49 This phenomenon is also consistent with our results that metabolites before GAPDH, G-5P/F-5P, F-1,6BP and DHAP, were highly accumulated in the cKO mice, suggesting that the cKO mice attempt to increase glycolytic metabolism to cope with the energy crisis.
Usually, cells including neurons will initiate a defensive mechanism after stress or gene deletion. We observed that Ido1, a critical gene in NAD+ de novo pathway is actually downregulated in the cKO mice. There are a couple of possibilities for the reduced expression of Ido1. First, our study was conducted 3 weeks after TAM administration. At this time point, the pathological conditions are fully developed, and other pathways may inhibit Ido1 expression. Second, Ido1 is indeed increased in early stage after NAMPT deletion, (e.g., one week after TAM administration), but we did not conduct time-dependent transcriptional profiling study. On the other hand, the NAMPT cKO mice eventually died, 17 indicating the rescue of other NAD+ biosynthesis pathways are not sufficient to prevent mouse death, further supporting that the NAD+ salvage pathway is the predominant pathway and NAMPT gene is essential for survival.
Up- and down-regulation of individual genes may only have a small influence on complex traits, the combined effects of genes within a signaling pathway have a greater possibility to affect complex phenotypes, and assessing the effects of gene expression in the context of pathways will help to understand phenotypic significance. Thus, we conducted GO and KEGG pathway analyses and GSEA, aiming to identify signaling pathways that have potential contributions to the pathological phenotype of the NAMPT cKO mice. These analyses revealed that pathways related to inflammation, immune response and cell death were upregulated while cellular function related pathways were downregulated. In GSEA of 4603 gene sets, we found that JAK-STAT, NF-κB, cytokine-cytokine receptor interaction and apoptosis signaling pathways were significantly activated in the cKO mice. The JAK-STAT pathway is activated by many cytokines and regulates cell growth, inflammation, and early embryonic development. 50 NF-κB pathway is involved in apoptosis, inflammation, cell survival and various autoimmune diseases. NF-κB induces the expression of various inflammatory pro-inflammatory genes, including those encoding cytokines and chemokines, participates in inflammasome regulation, and regulates the cell proliferation and apoptosis. 51 The complement system is a proteolytic cascade and mediates innate immunity and neuroinflammation. 52 One of the main consequences of complement activation is the recruitment of inflammatory and immunocompetent cells. Upregulation of these pathways in the cKO mice is consistent with our observation that the cKO mice exhibited enhanced reactive gliosis and motor neuron degeneration. 17 It is also consistent with results in human trials, where oral administration of NR has anti-inflammatory properties in muscle. 53 Since NAMPT is deleted in the projection neurons, this observation also indicates a non-cell autonomous effect of neuronal NAMPT on glial cell activation, a common phenomenon in brain injury and age-related neurodegenerative diseases including Alzheimer’s disease (AD). These pathways interact with each other, and activations of them may synergistically contribute to inflammation and cell death. On the other hand, function-related pathways including glutamatergic synapse, GABAergic synapse, cholinergic synapse, ion channel function and ion transportation activity were significantly down-regulated in the cKO mice, suggesting neuronal/synaptic functions were impaired, which likely affects their downstream pathways of signal transduction.
There are a few weaknesses in the current study. One is that we used brain tissues for metabolomic and transcriptional profiling while NAMPT was only deleted in neurons. Therefore, the results should reflect an overall response from multiple cell types. This might be particularly true when the brain tissues were collected ∼3 weeks after TAM injection when the pathological conditions were fully developed. Indeed, we observed that some astrocyte- and microglia-specific genes were highly upregulated in the cKO mice, suggesting a non-cell autonomous effect on reactive gliosis caused by neuronal NAMPT deletion. However, our results from cultured neurons confirmed the important role of neuronal NAMPT in energy production and bioenergetic homeostasis. The other weakness is that due to the nature of in vivo experiments with relatively small sample size, we analyzed data from mice of both genders. Both animal and human studies indicate that sex may affect protein expression and metabolism under normal and pathological conditions.54–56 However, there is no difference in brain weight between WT and the cKO mice in our study. 17 Age is another factor that may affect gene expression and metabolism. NAD+ metabolome is decreased with age.57–59 Transcriptomic analysis also suggests impaired microglia response to cerebral ischemia in aged mice compared with young mice. 60 In the current study, since there is a dramatic change in phenotype after NAMPT deletion, 17 the effect of NAMPT deletion on metabolism and other pathways should supersede other factors including age and sex, and thus the relative effect of NAMPT deletion is likely the same between male and female, and young and aged mice.
In summary, from our comprehensive study of combined metabolomic and transcriptional profiling, we conclude that NAD+ and NAD+ metabolome reduction, a direct consequence of NAMPT deletion, leads to enhanced energy depletion and oxidative stress, suppresses cellular function, and activates cellular processes related to apoptosis, immune response and inflammation, and eventually causes animal death. These processes are likely time dependent as the NAD+ contents in the brain tissue are progressively reduced following NAMPT deletion. 17 As NAD+ decline has been found in aging and many acute and neurodegenerative diseases, our current study provides metabolic and molecular insights into the mechanism of neurodegeneration and potential therapeutic strategies under these pathological conditions.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X21992625 - Supplemental material for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X21992625 for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT by Samuel Lundt, Nannan Zhang, Jun-Liszt Li, Zhe Zhang, Li Zhang, Xiaowan Wang, Ruisi Bao, Feng Cai, Wenzhi Sun, Woo-Ping Ge and Shinghua Ding in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-xlsx-2-jcb-10.1177_0271678X21992625 - Supplemental material for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT
Supplemental material, sj-xlsx-2-jcb-10.1177_0271678X21992625 for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT by Samuel Lundt, Nannan Zhang, Jun-Liszt Li, Zhe Zhang, Li Zhang, Xiaowan Wang, Ruisi Bao, Feng Cai, Wenzhi Sun, Woo-Ping Ge and Shinghua Ding in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-xlsx-3-jcb-10.1177_0271678X21992625 - Supplemental material for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT
Supplemental material, sj-xlsx-3-jcb-10.1177_0271678X21992625 for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT by Samuel Lundt, Nannan Zhang, Jun-Liszt Li, Zhe Zhang, Li Zhang, Xiaowan Wang, Ruisi Bao, Feng Cai, Wenzhi Sun, Woo-Ping Ge and Shinghua Ding in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-xlsx-4-jcb-10.1177_0271678X21992625 - Supplemental material for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT
Supplemental material, sj-xlsx-4-jcb-10.1177_0271678X21992625 for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT by Samuel Lundt, Nannan Zhang, Jun-Liszt Li, Zhe Zhang, Li Zhang, Xiaowan Wang, Ruisi Bao, Feng Cai, Wenzhi Sun, Woo-Ping Ge and Shinghua Ding in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-xlsx-5-jcb-10.1177_0271678X21992625 - Supplemental material for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT
Supplemental material, sj-xlsx-5-jcb-10.1177_0271678X21992625 for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT by Samuel Lundt, Nannan Zhang, Jun-Liszt Li, Zhe Zhang, Li Zhang, Xiaowan Wang, Ruisi Bao, Feng Cai, Wenzhi Sun, Woo-Ping Ge and Shinghua Ding in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-xlsx-6-jcb-10.1177_0271678X21992625 - Supplemental material for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT
Supplemental material, sj-xlsx-6-jcb-10.1177_0271678X21992625 for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT by Samuel Lundt, Nannan Zhang, Jun-Liszt Li, Zhe Zhang, Li Zhang, Xiaowan Wang, Ruisi Bao, Feng Cai, Wenzhi Sun, Woo-Ping Ge and Shinghua Ding in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-xlsx-7-jcb-10.1177_0271678X21992625 - Supplemental material for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT
Supplemental material, sj-xlsx-7-jcb-10.1177_0271678X21992625 for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT by Samuel Lundt, Nannan Zhang, Jun-Liszt Li, Zhe Zhang, Li Zhang, Xiaowan Wang, Ruisi Bao, Feng Cai, Wenzhi Sun, Woo-Ping Ge and Shinghua Ding in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-xlsx-8-jcb-10.1177_0271678X21992625 - Supplemental material for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT
Supplemental material, sj-xlsx-8-jcb-10.1177_0271678X21992625 for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT by Samuel Lundt, Nannan Zhang, Jun-Liszt Li, Zhe Zhang, Li Zhang, Xiaowan Wang, Ruisi Bao, Feng Cai, Wenzhi Sun, Woo-Ping Ge and Shinghua Ding in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-xlsx-9-jcb-10.1177_0271678X21992625 - Supplemental material for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT
Supplemental material, sj-xlsx-9-jcb-10.1177_0271678X21992625 for Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT by Samuel Lundt, Nannan Zhang, Jun-Liszt Li, Zhe Zhang, Li Zhang, Xiaowan Wang, Ruisi Bao, Feng Cai, Wenzhi Sun, Woo-Ping Ge and Shinghua Ding in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Health [National Institute of Neurological Disorders and Stroke (NINDS) grants R01NS069726 and R01NS094539 to SD] and the America Heart Association [Midwest Affiliate Grant-in-Aid (13GRNT17020004), and NCRG-IRG 16IRG27780023 to SD].
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
SL conducted experiments and analyzed data, and helped to edit manuscript draft. NZ conducted experiments. JL analyzed data. ZZ conducted experiments and analyzed data. LZ, XW and RB conducted experiments. FC analyzed data. WS analyzed data. WPG conducted experiments and manuscript edit. SD conceived and supervised the project, designed experiments, conducted experiments, analyzed data, wrote the manuscript, and acquired funding for the project. All authors read and approved the final manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.