
Abstract
Small non-coding RNAs (sRNAs) regulate the synthesis of virulence factors and other pathogenic traits, which enables the bacteria to survive and proliferate after host infection. While high-throughput sequencing data have proved useful in identifying sRNAs from the intergenic regions (IGRs) of the genome, it remains a challenge to present a complete genome-wide map of the expression of the sRNAs. Moreover, existing methodologies necessitate multiple dependencies for executing their algorithm and also lack a targeted approach for the de novo sRNA identification. We developed an Isolation Forest algorithm-based method and the tool Prediction Of sRNAs using Isolation Forest for the de novo identification of sRNAs from available bacterial sRNA-seq data (http://posif.ibab.ac.in/). Using this framework, we predicted 1120 sRNAs and 46 small proteins in Mycobacterium tuberculosis. Besides, we highlight the context-dependent expression of novel sRNAs, their probable synthesis, and their potential relevance in stress response mechanisms manifested by M. tuberculosis.
Introduction
Tuberculosis is caused by Mycobacterium tuberculosis, which infects about 10 million people per year. 1 The pathogenicity of M. tuberculosis depends on its ability to adapt to the changing host environment and transmit a signal to subvert the host immune response to maintain latent infection. As a defense mechanism, the host immune system hinders the growth of bacteria by imposing a variety of stresses such as oxidative stress, acidic pH, and nitrogen stress. 2 M. tuberculosis responds to these stress conditions by modulating gene regulation and revised expression of certain genes across the genome.
Regulatory proteins, small non-coding RNAs (sRNAs), and their target genes constitute gene regulatory networks, enabling bacteria to adapt to their metabolic needs and express virulence factors at different stages of infection in a coordinated manner. 3 Bacterial sRNAs can influence the expression of genes across various stages of genetic information flow, encompassing transcription and translation processes. A variety of mechanisms are used by sRNAs to regulate translation and mRNA stability, including changes in RNA conformation, base pairing with target mRNA, and interaction with proteins.4-6 Based on base pairing with their target mRNAs, sRNAs are broadly classified into the following categories (i) Antisense or cis-encoded sRNAs, which are present on the complementary strand to Open Reading Frames (ORFs), 5’ or 3’ untranslated region (UTR) of an mRNA and share an extended region of complete complementarity with their target and (ii) Trans-encoded sRNAs which act in trans on distant targets. These sRNAs share limited complementarity with their target and are largely found in the IGRs.4-6
Most of the previously described sRNAs are exclusively encoded within IGRs. However, recent studies indicate their presence in gene UTRs, which can be derived via endoribonuclease-mediated mRNA degradation-based pathways.7-10 For example, the cpxP membrane stress response chaperone is transcribed into an mRNA containing a conserved 60-nucleotide 3’-UTR, which can independently act as an Hfq-dependent sRNA. This sRNA, CpxQ, is excised from the mRNA by RNase E, enabling its activity as a regulator of multiple genes. 7 Another study in E. coli reported a similar mechanism wherein sRNAs are carved out of protein-coding regions, rather than from 3’-UTR or 5’-UTRs, during mRNA decay. 11 In some cases, premature transcription termination can also give rise to sRNAs located in 5’-UTR, while in others, 5’-UTR riboswitch acts as a non-coding sRNA and regulates the expression of distantly located target mRNA.7,12 Also, the “junk RNA” derived from riboswitches and mRNAs has been proven to be functionally important as small non-coding RNAs. 13
Currently, available tools utilize comparative genomics, expression data analysis, secondary structure comparison, and transcription signal-based methods to identify sRNAs in bacterial genomes.14,15 Tools such as QRNA 16 and Intergenic Sequence Inspector 17 focus on sRNAs conserved within the IGR among related genomes. Secondary structure-based prediction consists of two approaches, namely, thermodynamic stability-based tools such as RNALfoldz 18 and structure consensus-based tools such as RNAz, which are dependent on the minimum free energy (MFE) of sRNA’s secondary structures. 19 Transcriptional signal prediction tools include sRNApredict, 20 sRNAscanner, 21 and sRNAfinder, 22 which use either expression-based signals or pre-computed coordinates such as orphan transcriptional signals in the IGRs or other predictive features such as promoter signals, transcription factor binding sites, or terminator signals as input for the prediction. Whereas the expression-based approach identifies sRNAs in the IGRs based on their expression compared to upstream and downstream protein-coding genes. 14
Recent advances in next-generation sequencing have offered a more versatile and reliable way of profiling sRNAs, hence providing a better understanding of bacterial transcriptomes including sRNAs. These studies have revealed the expression of small transcripts from 5’ or 3’-UTRs of mRNAs or internal fragments of mRNAs, rRNAs, or tRNAs.9,23 As sRNAs can be produced from 3’-UTRs, 5’-UTRs, internal RNA fragments of mRNAs, or as independent transcripts with the help of alternate promoters, demarcation of these regions from RNA sequencing data can be challenging. Tools such as Analysis of Paired-End RNA-Seq Output (APERO) and sRNA-Detect use RNA-seq data and predict sRNA based on the enrichment of read starts and genomic stretch with constant read coverage, respectively.24,25 While RNA-seq provides a comprehensive view of the entire transcriptome, sRNA-seq focuses on sRNAs, typically those in the range of 20 to 200 nucleotides. Therefore, sRNA-seq allows for a targeted analysis of the sRNA population, which might be overlooked or underrepresented in total RNA-seq data.
sRNA-seq technology involves direct cloning and massively parallel sequencing by synthesis that allows the discovery of sRNAs and small regulatory proteins across the entire bacterial genome. 26 Some of the sRNA sequence analysis tools such as seqpac and sRNAPipe are designed primarily for eukaryotes, and either employ sequence-based counting methods or mapping-based methods to discern small transcripts.27,28 While such methods for analyzing sRNA-seq data exist, an effective method for de novo sRNA identification from sRNA-seq data is not yet available for bacteria.
We conducted a global screen of sRNAs in M. tuberculosis H37Rv using the Isolation Forest algorithm implemented on publicly available sRNA-seq data. 29 Isolation Forest has been used to identify invasive alien species (outliers) in biological geo-profiling 30 and genomic islands based on genomic patterns. 31 This algorithm isolates anomalous data points, that is, outliers from a given data by leveraging a random partitioning strategy. The data is iteratively split into two subsets using randomly chosen values as the threshold within the current range, eventually forming a large binary tree. In this process, anomalous data points are more likely to be separated earlier and take less splitting operation compared to a normal data point. Assuming the path length of each data point stands for the number of splits required to separate the data point from the rest of the subset, we calculated the anomaly score for each of the data points as the average path length calculated from multiple binary trees constructed from the same data. Therefore, more anomalous points will have less anomaly scores and vice versa. In our study, we implemented this technique on per-base read count data, where the abnormally higher read counts (anomalous points) signify data points corresponding to sRNA expression. Additional steps such as grouping consecutive anomalous points and length filtration were considered for precision in identifying sRNA coordinates. 32 Through this approach, we report 1120 sRNAs and 46 small proteins across 6 diverse growth conditions. Many of these sRNAs in both protein-coding and non-protein-coding regions appear important for M. tuberculosis adaptation to host stress environments. This machine learning-based method operates on per-base coverage obtained from sRNA-seq data to extract potential sRNA coordinates with significant expression, thereby enabling a targeted and genome-wide identification of sRNAs. Based on this method, we developed Prediction Of sRNAs using Isolation Forest (POSIF), a tool for de novo bacterial sRNA identification. Unlike previously available tools, POSIF has a user-friendly interface and doesn’t have external dependencies.
Methods
Data retrieval and processing
sRNA-seq data for six different growth conditions with accession number SRP142345 were retrieved from NCBI-Sequence Read Archive (NCBI-SRA) (https://www.ncbi.nlm.nih.gov/sra) (Table S1). 29 These SRA files were converted to fastq files using sra-toolkit fastq-dump (version 2.10.0). The adapter sequences and reads less than 20 nucleotides in length were trimmed using Trim-Galore for each of the six growth conditions (version 0.6.2). 33 Trimmed reads were aligned to the M. tuberculosis H37Rv (NC_000962.3) reference genome using Bowtie2 (version 2.3.5.1). 34 Reads mapped to the reference genome were separated into forward and reverse-strand Binary Alignment and Map (BAM) files, which were further sorted and indexed using Samtools (version 0.1.9). 35 Per-base coverage was computed for each strand using Bedtools (version 2.26.0). 36
Prediction of putative sRNA regions using Isolation Forest
The Isolation Forest algorithm is designed to detect and isolate anomalous data points by assigning them an anomaly score. This score represents the average depth at which each data point is isolated within a collection of decision trees. We implemented the Isolation Forest algorithm 32 from the Python package scikit-learn (0.22.1) and used it to predict putative sRNA regions from the processed sRNA-seq data. The data included rich medium, acidic pH, iron limitation, membrane stress, nutrient starvation, and oxidative stress growth conditions (SRP142345). 29 Per-base coverage files in .bed format were given as input to the program, along with the contamination factor parameter set as 0.005 (i.e., 0.5% outlier). Consecutive points were considered as peaks and two peaks separated by ⩽5 nucleotides were merged to derive a single sRNA expression region. Regions with < 20 nucleotides were not considered for further analysis.
To represent the context-dependent expression of the predicted sRNAs, the offset score, which depicts the contamination factor percentile of the inverse anomaly score, was used as a benchmark expression level in each condition.
Annotation and classification of predicted regions
The final set of non-redundant sRNAs across growth conditions was derived by merging regions if they overlap by > 75% of the length. From these, regions mapping to known repeat regions, tRNAs, and rRNAs were removed. The final list comprised 1166 putative sRNAs, of which 46 were marked as small proteins of > 10 amino acid length with a start codon and in-frame stop codon. The genomic location of these predicted sRNAs and small protein regions was identified by mapping the midpoint of these regions to annotated genes, UTRs, and IGRs in M. tuberculosis.
Genomic coordinates of protein-coding genes, tRNA, and rRNA were derived from the NCBI RefSeq gene annotation file (https://www.ncbi.nlm.nih.gov/refseq/). 37 Transcription termination (3’-UTR) coordinates were obtained from the WebGeSTer database. 38 Coordinates of transcription start sites (5’-UTR) were downloaded from Cortes et al. 39
Conservation of sRNAs and small proteins
To test the conservation of identified sRNAs across mycobacterial species and other bacteria, a blastn-short search specific for short nucleotide segments was performed using BLASTN version 2.6.0 with the E-value cutoff of 1E−04 and all other default parameters. 40 The target database was a custom-made offline database that included the following genomes: Gram-negative bacteria (Escherichia coli K-12, Haemophilus influenzae Rd KW20, Klebsiella pneumoniae HS11286, Pseudomonas aeruginosa PAO1, Vibrio cholera N16961, and Yersinia pestis CO92), Gram-positive bacteria (Bacillus subtilis 168, Listeria monocytogenes EGD-e, Staphylococcus aureus NCTC 8325, Staphylococcus epidermidis ATCC 12228, Streptococcus mutans UA159, and Streptococcus pneumoniae R6), and mycobacteria (Mycobacterium africanum GM041182, Mycobacterium canettii CIPT 140010059, Mycobacterium smegmatis FDAARGOS_679, Mycobacterium leprae TN, Mycobacterium bovis AF2122/97, Mycobacterium avium 104, Mycobacteroides abscesses ATCC 19977, Mycobacterium haemophilum DSM 44634, and Mycobacterium marinum E11).
Small protein conservation was tested using BLAST-p against the NCBI-nr database. From the Conserved Domain Database (CDD) of NCBI, CD-Search was used to identify the conserved domains in the small proteins. 41 The MFE for the 1120 predicted sRNAs, 65 experimentally validated sRNAs in M. tuberculosis, and 1120 randomly chosen IGRs were calculated using RNA-fold software. 42 P-values were calculated using the Wilcoxon rank sum test implemented in R (https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/wilcox.test).
Alternative transcription starts site (TSS) and RNase E-mediated synthesis of sRNAs
To verify sRNA expression, the start sites of the predicted regions were compared with genome-wide TSS identified in M. tuberculosis in rich medium and nutrient starvation growth conditions.39,43 The TSS of a predicted sRNA was validated if: (a) the TSS site is located within 10 bp upstream or downstream of the sRNA start site, (b) the sRNA is located between a given gene and its annotated TSS, and (c) the sRNA is close to a reported alternate TSS.
To check whether sRNAs were synthesized via RNase E-mediated mRNA decay, RNA-seq data for RNase E mutant (SRR8550314, SRR8550315, and SRR8550316) and wild type (SRR8550302, SRR8550303, and SRR8550304) samples from the rich medium of M. tuberculosis were analyzed. 44 The SRA files were downloaded from NCBI-Sequence Read Archive (NCBI-SRA) (https://www.ncbi.nlm.nih.gov/sra) and converted to .fastq files using sra-toolkit fastq-dump (version 2.10.0). 45 The adapter sequences and reads less than 20 nucleotides in length were trimmed using Trim-Galore (version 0.6.2) (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) 33 for each replicate of the two growth conditions. Trimmed reads were aligned to the M. tuberculosis H37Rv (NC_000962.3) reference genome using Bowtie2 (version 2.3.5.1). 34 The BAM files generated for three replicates per growth condition were merged using Samtools (version 0.1.9) 35 to obtain a single BAM file each for RNase E mutant and wild type. Per-base coverage files for each of the growth conditions were generated from the respective BAM file using Bedtools (version 2.26.0). 36 The per-base files were used as input to an in-house Python script to identify sRNA regions that show decreased expression in RNase E mutant growth conditions compared to the wild type. The significant difference (adjusted P-value < 0.05) in the expression of sRNAs was computed using the Wilcoxon rank sum test and Student’s T-test, 46 based on the difference in the read count distribution of the respective region between the RNase E mutant and the wild-type samples.
Gene targets of the identified sRNAs
Potential gene targets of the identified sRNAs were predicted using TargetRNA2. 47 The reference genome of M. tuberculosis H37Rv (NC_000962.3) was used as the input file. For each sRNA, gene targets were generated based on the binding energy and P-value. RNA-Seq data from M. tuberculosis for multiple growth conditions such as acidic pH 4.5 (SRR10115602), oxidative stress (SRR13019063), nutrient starvation (ERR262987), membrane stress (SRR17730393), iron limitation conditions (SRR1917709), and rich medium (ERR262983) were retrieved to quantify the expression of the genes with FDR ⩽ 0.05. 48
The SRA and GSM files were downloaded from NCBI-SRA (https://www.ncbi.nlm.nih.gov/sra), and the paired-end sequence was then split into forward and reverse sequences using sra-toolkit fastq-dump (version 2.10.0). For each growth condition, one of the replicates was chosen based on FastQC reports for further analysis. In the instances where multiple replicates exhibited similar FastQC profiles, a replicate was selected based on greater mean depth and mean coverage provided in the Samtools coverage report. The adapter sequences and reads less than 20 nucleotides in length were trimmed using Trimmomatic (version 0.39) for all growth conditions. 49 Trimmed reads were aligned to the M. tuberculosis H37Rv (NC_000962.3) reference genome using Bowtie2 (version 2.4.5). 34 Per-base coverage was generated for the respective BAM files using Bedtools (version 2.30.0) 36 and Fragments Per Kilobase of transcript per Million (FPKM) normalization was performed to quantify the expression of genes. For each growth condition, genes with expression over the median value were marked as highly expressed while the ones below the median were marked as less expressed.
Prediction Of sRNAs using Isolation Forest
Prediction Of sRNAs using Isolation Forest predicts bacterial sRNA regions using sRNA-seq data. The tool accepts per base .bed file (strand-separated or non-strand-separated), contamination factor (percentage of anomalous data), and the organism’s name. Prediction Of sRNAs using Isolation Forest utilizes Isolation Forest, which is an unsupervised machine learning algorithm from the Python package scikit-learn (0.23.1) that detects outliers (or anomalies) from large data based on the anomaly score assigned to each point in the data. 50 The algorithm builds an ensemble of decision trees (iTrees) that contain anomalies isolated closer to the root of the tree and normal instances at the deeper end of the trees. 32 Currently, POSIF can be run for 10 bacteria, namely, Escherichia coli K-12, Salmonella enterica serovar Typhimurium LT2, Pseudomonas aeruginosa PAO1, Staphylococcus aureus NCTC 8325, Bacillus subtilis 168, Vibrio cholerae, Listeria monocytogenes EGD-e, Clostridium difficile, Mycobacterium tuberculosis H37Rv, and Streptococcus pneumoniae Hu17. The back-end of POSIF is coded in Python and utilizes libraries Celery, Flask, Scikit-Learn, Pandas, and Redis. The output generated by the tool is a downloadable ZIP file that includes predicted sRNAs and their respective genomic locations in a .csv format. Performance of POSIF was compared with APERO, which predicted a total of 148399 regions with lengths ranging from 15 to 12768 nucleotides. We further filtered these results based on (a) the threshold value, that is, (20×total number of reads)/genome size in column “freq” (reads at the start position) (Leonard et al), 24 (b) Fstart, E-value (coverage ratio of the last position of the transcript), using a threshold value of 0.0538, (c) merged sRNAs with at least 75% overlap from both side or 100% from one side, and (d) length range of 20–250 nucleotides, resulting in 1373 sRNAs. 24
All statistical tests were performed using Python and R statistical packages.50-55 All data were analyzed using in-house Python and R scripts. Classification and line plots were generated using the Python library Matplotlib 3.2.1.
Results
M. tuberculosis sRNA-repertoire identified using Isolation Forest includes many novel and conserved sRNAs
Multiple systemic networks in bacteria are regulated by sRNAs in response to environmental conditions. Here, we used sRNA-seq data from a previously published study by Gerrick et al 29 for the genome-wide detection of such regulatory sRNAs using the Isolation Forest machine learning algorithm. 32 For the sRNA-seq data, outliers detected by the Isolation Forest algorithm are the highly expressed (high coverage) base positions and have lower anomaly scores. When outliers are detected in consecutive order, they are treated as sRNA expression signals (peaks) (Figure S1). We utilized M. tuberculosis sRNA-seq data encompassing five stress conditions (acidic pH, iron limitation, membrane stress, nutrient starvation, and oxidative stress) and a rich medium growth condition for the sRNA prediction (Table S1). We identified a total of 1166 potential sRNA-encoding regions with a median length of 87 nucleotides expressed in one or more of these growth conditions (Table S2) (Figure S2). We successfully captured 21 out of the 65 experimentally verified sRNAs in M. tuberculosis (Table S3). For instance, MTS2823 captured as ncRv3661i in our data was expressed in all the studied growth conditions (Figure S3a). 56 Also, MrsI, which is a known sRNA involved in the regulation of gene expression during iron limitation in M. tuberculosis, was captured as ncRv1847u. 29 Our method captures MrsI as expressed in iron limitation condition, membrane stress, and oxidative stress conditions (Figure S3b). sRNA B55 was identified in our data as ncRv0609u, which showed expression in all the growth conditions (Figure S3c). 57 Further, we compared our predictions with previously published works to underscore the importance of our findings. We captured 39 sRNAs that had previously been identified in the IGRs using a moving-window approach in M. tuberculosis. 13 Also, 89 of the sRNAs identified in our study were previously described by Wang et al. 58 A comparison of the predicted sRNAs with 189 sRNAs identified earlier using the BS_Finder tool in M. tuberculosis showed 117 as common (Table S4). 29 Accurate capturing of some of the known sRNAs, therefore, underscores the utility of our method in predicting sRNAs using high-resolution sRNA-seq data.
Further, we studied the conservation of predicted sRNAs across mycobacteria and other Gram-positive and Gram-negative bacteria (Table S5). We observed that 1111 of the sRNAs were conserved across all the mycobacterial species analyzed, while 1115 sRNAs were selectively conserved in Mycobacterium bovis and Mycobacterium leprae. Interestingly, about 355 sRNAs were present in the rapidly growing mycobacteria (Mycobacterium abscessus and Mycobacterium smegmatis) compared to slowly growing mycobacteria. We observed that 27 sRNAs were conserved in mycobacteria as well as Gram-negative bacteria. These findings suggest the evolutionary significance and potential functional importance of sRNAs in diverse bacterial species.
Understanding the sRNA folding kinetics and MFE is an important aspect of assessing the accuracy and reliability of the predicted sRNAs. We compared the structural stability (MFE of their secondary structures) of the sRNAs against 65 experimentally validated sRNAs in M. tuberculosis. Along with these, 1120 randomly selected IGRs were included as the negative control. We observed that the MFE of the sRNAs is significantly lower than the random IGRs, suggesting their stability (P-value = 2.33e−26; Figure 1). Beyond this, we studied if any of the identified sRNAs are bound by start and stop codons in-frame. We identified 46 putative small proteins, 10 of which had the TSS identified in their upstream regions (see “Methods” section, Table S6).39,43 For instance, MTB_sORF_38 identified as a putative upstream ORF of operon Rv3001c-Rv3003c has a nearby TSS (Figure 2). Also, our search in the non-redundant Refseq protein database uncovered conserved domains in putative small proteins (Table S6). For example, MTB_sORF_11 with a PPE domain (with Pro-Pro-Glu motif) is expressed during membrane stress and nutrient starvation (Figure S4). In addition, among 324 annotated proteins < 100 amino acids in the NCBI database, 31 sRNAs exhibited length overlap of more than 90% (Table S7).
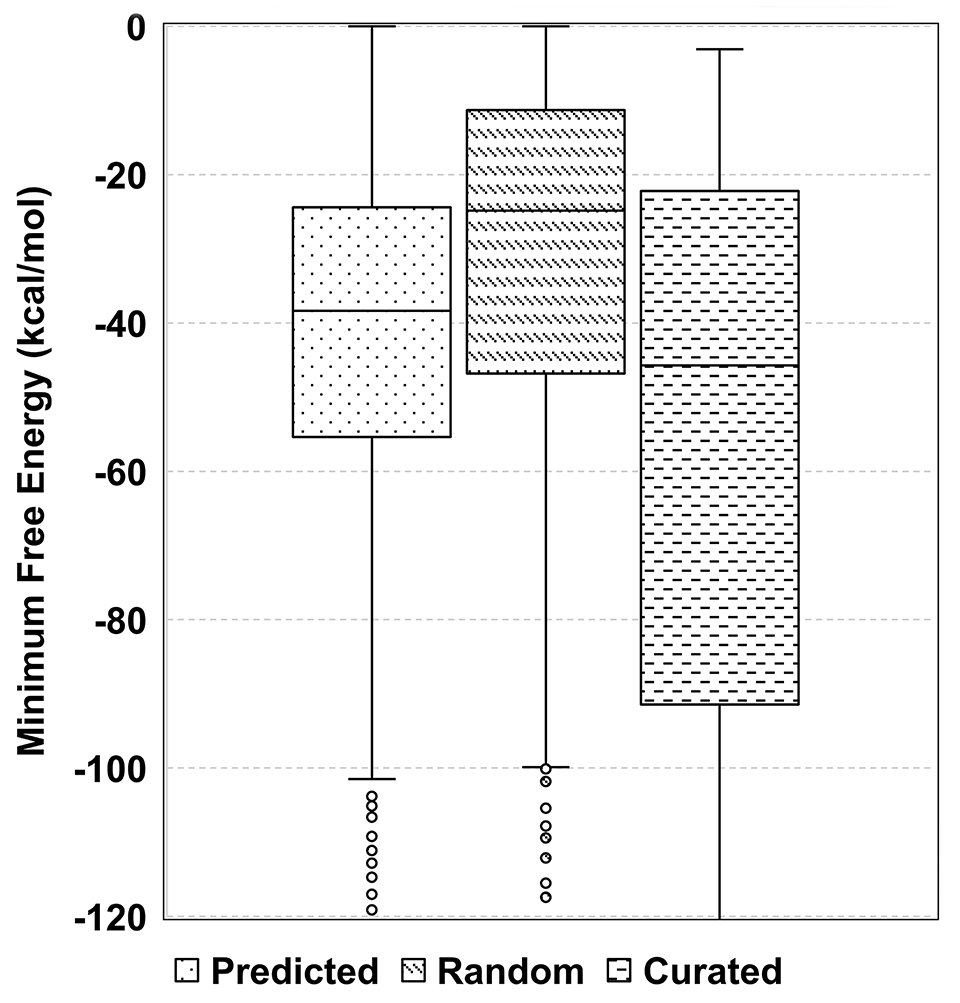
Structural stability of the sRNAs. Box plot comparing the structural stability of 1120 predicted sRNAs, 65 curated sRNAs, and 1120 randomly selected genomic sequences from IGRs with comparable lengths. Wilcoxon ranked sum test has been performed between Random and Predicted sRNAs (P-value = 2.33e−26), and Predicted and Curated sRNAs (P-value = 0.0325)

Expression of MTB_sORF_38. The putative small protein MTB_sORF_38 encoded on the negative strand of the genome shows expression across all the studied growth conditions. Anomaly scores are plotted on the negative Y-axis, read counts are on the positive Y-axis, and the genomic position is represented on the X-axis. The prominent red bold line corresponds to MTB_sORF_38, and the adjacent blue dotted line is the nearby TSS.
Context-dependent expression of the sRNAs suggests their importance in mediating M. tuberculosis stress response
sRNAs are the crucial part of the regulatory network, which are strongly induced during stress conditions and regulate a set of targets to adapt to various stress conditions imposed by the host tissue.2,3 We observed that while 5.89% (66/1120) sRNAs showed expression irrespective of the growth condition, certain sRNAs were expressed in a particular stress condition, a combination of stress conditions, or only in the rich medium (Table S8). For instance, sRNA ncRv3825 F which is encoded sense to Rv3825c showed significant expression in all the growth conditions (Figure 3) (Table S9). This sRNA is conserved across mycobacteria, including M. africanum, M. canettii, M. bovis, and M. avium. One of the potential targets predicted for this sRNA is Rv3009c, which codes for glutamyl-tRNA(gln) amidotransferase subunit B (gatB) (see “Methods” section). gatB is essential for the growth of bacteria 59 and shows high expression in all the growth conditions, which could be a result of positive regulation by ncRv3825 F (Figure S5). About 10.44% (117/1120) of the total sRNAs are expressed only in the rich medium. For instance, sRNA ncRv3547i expressed under a rich medium is predicted to target a gene encoding a conserved protein Rv2410c (Figure S6a). Interestingly, Rv2410c is downregulated exclusively under growth in rich medium (Table S10). The binding of ncRv3547i to the target gene covers its start codon, potentially impeding proper binding or movement of the ribosomes (Figure S6b). Therefore, we speculate that sRNA ncRv3547i negatively regulates Rv2410c in rich medium.

Expression of ncRv3825F under all the growth conditions. sRNA ncRv3825F is highly expressed across all the investigated growth conditions. The expression levels are represented on the positive Y-axis (red line), while the negative Y-axis displays the anomaly score. The yellow dotted line on the negative Y-axis denotes the offset score associated with each growth condition (see “Methods” section).
We observed that many sRNAs showed stress-dependent expression. sRNAs ncRv0983B, ncRv2711B, ncRv2839B, and ncRv3616 are expressed across all the stress conditions, which marks their importance in general stress response in M. tuberculosis (Table S2). Notably, sRNAs ncRv2711B and ncRv2839B are predicted to target Rv0827c (kmtR), which is involved in oxidative stress response by detoxification of host-generated free radicals. 60 In addition, one of the targets predicted for ncRv0983B is Rv2911 (dacB2), which contributes to cell wall permeability and integrity under stress. 61 Rv0017c encodes cell division protein RodA, which is a potential target for ncRv0983B. Rv0017c is downregulated in iron limitation, Nutrient starvation, and acidic pH. Also, about 12.32% (138/1120) of the sRNAs were expressed only in acidic pH, 10% (112/1120) were expressed only in iron limitation, 5.35% (60/1120) were expressed only in membrane stress, 8.48% (95/1120) were expressed in nutrient starvation and 10.44% (117/1120) were expressed in oxidative stress (Figure S7). Below, we provide examples illustrating the stress-specific expression of sRNAs and their putative targets:
a. sRNA ncRv0140 expressed only in acidic pH is predicted to target Rv2626c (hrp1), which is a hypoxic response protein (Figure 4). hrp1 is downregulated in the acidic pH condition (Table S10). The binding of ncRv0140 is predicted at the start site or ribosome binding site of Rv2626c, which could result in the negative regulation of the putative target gene (Figure S8).
b. ncRv0188B is exclusively expressed during nutrient starvation (Figure S9a). Its potential targets, Rv3920c and Rv3628, are downregulated under nutrient starvation. Rv3920c codes for Jag protein crucial for cell division, and Rv3628 (ppa) encodes inorganic pyrophosphatase essential for intermediary metabolism and respiration. ncRv0188B is predicted to bind to the start site of these target genes (Figure S9b), which suggests the downregulation of Rv3920c and Rv3628 during nutrient starvation.
c. sRNA ncRv0188A expressed under nutrient starvation and predicted to target Rv2394 (ggtB) which exerts a key role in the gamma-glutamyl cycle (Figure S10a). Furthermore, it counteracts the surplus glutathione within the cells. By cleaving glutathione (GSH), GgtB produces a bactericidal dipeptide, underscoring the significance of ggtB downregulation for bacterial survival.62,63 Notably, ggtB is downregulated during nutrient starvation. We propose that binding of ncRv0188A to the start site of ggtB could interfere with the ribosome movement, thus negatively regulating ggtB under nutrient starvation (Figure S10b).
Therefore, further experimental analyses could reveal how the bacteria utilize these sRNAs to regulate gene expression in different growth conditions to withstand the hostile environment imposed by the host.

Context-dependent expression of ncRv0140. sRNA ncRv0140 is highly expressed only under acidic pH growth conditions. This sRNA overlaps with the gene Rv0140, coding for a conserved protein. Red lines on the positive Y-axis and negative Y-axis display expression levels as read counts and anomaly scores, respectively. The offset score for each growth condition is represented as a yellow dotted line on the negative Y-axis.
Many M. tuberculosis sRNAs are potentially generated via alternate TSS or RNase E-mediated degradation
The bacterial genome harbors a large set of sRNAs, which could be synthesized via multiple mechanisms based on its position on the genome.11,39 Therefore, we annotated and classified the predicted sRNAs based on their genomic position with respect to the annotated genes, UTRs, and IGRs. Our analysis discovered 874 mRNA subsegments, 805 cis-encoded, 69 trans-encoded (including UTRs), and 84 absolute IGR (excluding known UTRs) encoded sRNAs. Over 71.88% of the predicted sRNAs significantly overlapped with the known protein-coding sequences. In about 62.85% of these cases, predicted sRNA was entirely carried in the protein-coding sequence, while in 9.01% of cases, predicted sRNA overlapped with the protein-coding sequence and extended till the UTR of the respective gene. The remaining 28.125% of predicted sRNAs overlapped with 5’-UTR or 3’-UTR of a known protein-coding sequence or the sequence antisense to a known protein-coding sequence, UTRs, and absolute IGRs (Figure S11).
Similar to protein-coding regions, sRNA transcription also requires a TSS upstream or at the start of the sRNA coding region. Therefore, to authenticate the expression of the predicted sRNAs in our data, we mapped the start sites of the sRNAs to the known genome-wide TSS in M. tuberculosis derived from exponential and nutrient starvation growth conditions.39,43 Of the predicted sRNAs expressed in rich medium, 32.63% (141/432) show TSS near the sRNA start site (detailed in “Methods” section). There are 245 sRNAs in a rich medium, which appear to be generated from the known protein-coding regions. Among these, 15.91% (39/245) sRNAs have a neighboring TSS, which includes 20 internal TSS (iTSS), 2 alternative TSS (2/39), and 17 Primary TSS (Table S11). For example, the start site of the sRNA ncRv3045 matched with an internal TSS of Rv3045 (Figure 5). ncRv3045 is conserved in the genomes of M. africanum, M. canettii, M. bovis, M. avium, M. haemophilum, M. leprae, and M. marinum. This sRNA showed high expression in all the studied growth conditions except in nutrient starvation. One of the putative targets of this sRNA is Rv1103c (mazE3), a component of the Rv1102c–Rv1103c toxin-antitoxin system in M. tuberculosis. These genes orchestrate reversible bacteriostasis, facilitating adaptation to adverse stress conditions. 64 Therefore, TSS study in other growth conditions will help explain the expression of the numerous other sRNAs across the M. tuberculosis genome. In another example, our analysis suggests that ncRv3581 is generated from the CDS (Coding Sequence) of essential gene Rv3581c, which is expressed under all growth conditions (Figure S12a). It appears that this sRNA is generated from an internal TSS situated inside the coding sequence of Rv3581c (Figure S12b).

TSS-driven synthesis of ncRv3045. sRNA ncRv3045 is located within the CDS of Rv3045 and is detected in all the studied growth conditions except in nutrient starvation. A previously annotated internal TSS (iTSS) located immediately upstream of the ncRv3045 (indicated by the blue vertical line) appears to be driving the transcription of the sRNA. Expression—red line on the positive Y-axis, anomalous score—red line on the negative Y-axis, offset score—yellow dotted line on the negative Y-axis.
Further, we hypothesized that some of the sRNAs in our data could be synthesized via RNase E-mediated mRNA decay. 11 For this, we utilized RNA-seq data of M. tuberculosis RNase E wild type and RNase E mutant samples. 44 Of the predicted sRNAs from the rich medium growth phase, 56.713% (245/432) overlapped with the known protein-coding regions, and the start site of 15.92% (39/245) of these regions mapped to the known TSS. We analyzed significant differences (adjusted P-value ⩽ 0.05) between the expression of the remaining regions 84.08% (206/245) in RNase E mutant and RNase E wild type. From these regions, 52.91% (109/206) showed significantly high expression in the RNase E wild type compared to the RNase E mutant (Table S12) (P < 8.908e−06). For example, ncRv0510 and ncRv3211A appear to be excised from the coding sequences of Rv0510 and Rv3211A, respectively, under the influence of RNase E-mediated mRNA decay (Figures 6A and B). The sRNA ncRv0510 showed expression in acidic pH and rich medium growth conditions and high conservation across M. bovis, M. canetti, and M. africanum, which are part of M. tuberculosis complex (MTBC) and also in M. abscessus, M. avium, M. haemophilum, M. leprae, and M. marinum (Figure S13a). fadD26 is identified as a probable target of ncRv0510 where the sRNA binding is predicted in the 5’ UTR region of fadD26. FadD26 is essential for the synthesis of the virulence factor phthiocerol dimycocerosates (DIM), which is crucial during the early stages of infection (Figure S13b).65,66 Also, the sRNA ncRv3211A showed expression in acidic pH, membrane stress, and rich medium growth phase (Figure S13c). This sRNA showed high conservation across members of MTBC and in M. abscessus, M. avium, M. haemophilum, M. leprae, and M. marinum.
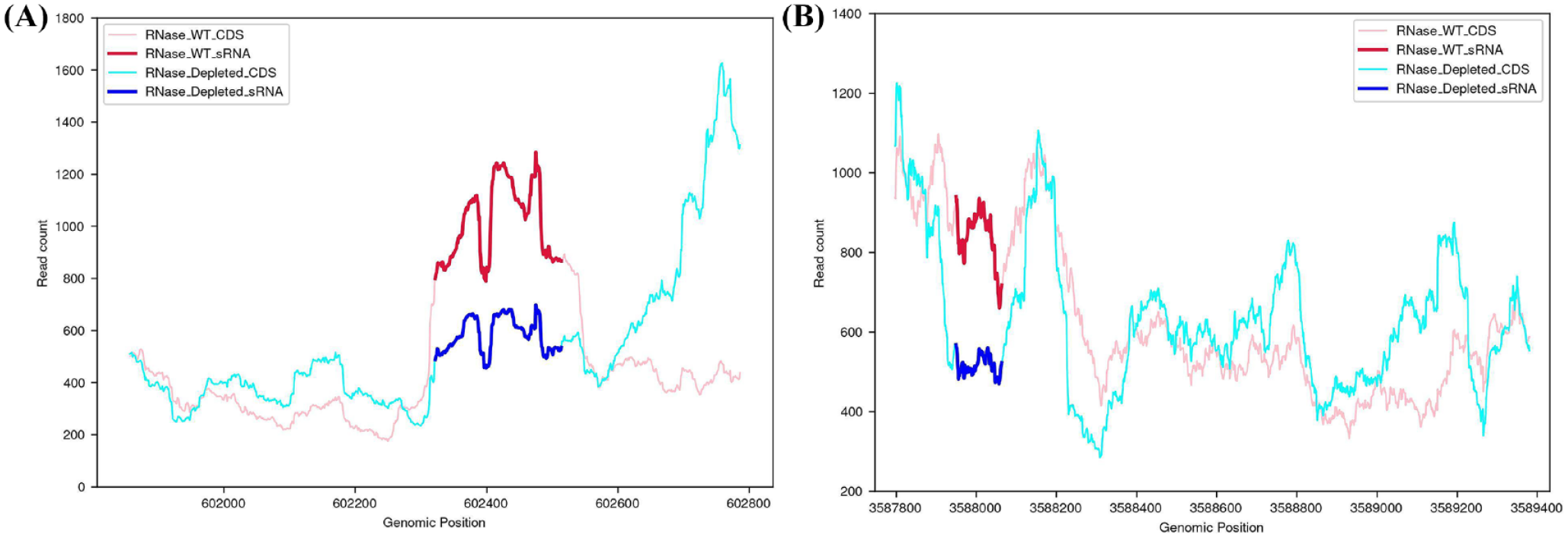
sRNA Expression in RNaseE wild type and depleted conditions. sRNA expression under RNase E wild type (red line) and RNase E depleted (blue line) conditions. Expression is quantified using RNA-seq data (see “Methods” section) (A) sRNA ncRv0510 and (B) sRNA ncRv3211. Significantly higher expression in the RNase E wild-type condition compared to RNase E depleted conditions suggests a possible RNase E-mediated mRNA decay mechanism behind the generation of these sRNAs.
In such findings, we observed that the expression throughout the gene was similar between RNase E wild type and mutant, except for the region predicted as putative sRNAs in our data. Collectively, these results suggest that the RNase E-mediated mRNA decay could be one of the crucial mechanisms for synthesizing many sRNAs in M. tuberculosis. We note that some of these mRNA subsegments could also be produced as intermediates during mRNA degradation without any functional relevance. 11 Further studies are needed to examine this aspect of sRNA synthesis and function.
Outlook of the tool P rediction O f s RNAs using I solation F orest
We developed our Isolation Forest-based method to detect sRNA regions from bacterial sRNA-seq data into a Graphical User Interface (GUI) tool POSIF (http://posif.ibab.ac.in/) using a Flask web application. The tool provides the option to predict sRNAs from the strand-specific or non-strand-specific bacterial sRNA-seq data. Outliers (read count as a feature) are detected at each base position from the per-base coverage file by the tool. The consecutive outliers are collectively considered as the sRNA expression signal/peak (detailed in Methods).
The output link generated by the tool is a downloadable .zip file that contains the predicted sRNA coordinates and their genomic location, which can be further analyzed to understand the functional relevance of these regions. We tested the performance of POSIF with other sRNA analysis tools. Currently available sRNA-seq analysis tools sRNAPipe and seqpac do not perform de novo sRNA detection.27,28 While the tools APERO and sRNA-Detect traditionally consider RNA-seq data as input, we ran them with sRNA-seq data (SRR7058126) and analyzed their results (Table S13). When tested with RNA-seq data (ERR262983), sRNA-Detect generated 18,487 sRNAs. 48 Running the tool on paired-end sRNA-seq data exceeded a run-time of 72 hours without producing any output files. Using APERO, we obtained 1373 sRNAs with lengths ranging from 20 to 250 nucleotides which included 10 experimentally verified sRNAs (see “Methods” section). Whereas in the rich medium, POSIF predicted a total of 432 sRNAs, of which it identified 13 that were experimentally validated. While the tool successfully processes both RNA-seq and sRNA-seq data, the run-time for the latter exceeds 12 hours, compared to 1 hour for RNA-seq data. Moreover, APERO is dependent on multiple external libraries, each requiring specific versions for the proper functionality, making it difficult to use. In contrast, POSIF runs on both single-end and paired-end sRNA-seq data, has a shorter run-time, is accessible online without any dependency, and generates quality output, including many experimentally validated sRNAs. Therefore, we believe that POSIF will be invaluable in sRNA-seq data analysis and in identifying novel sRNAs in bacteria.
In conclusion, our method for identifying sRNAs has yielded a set of promising candidates for further studies. Some of the potential sRNAs we described show distinct expression patterns, suggesting their context-dependent functions. Further experimental studies will provide insights into their functions and their role in M. tuberculosis pathogenicity. In addition, the current study provides a valuable tool for identifying and prioritizing bacterial sRNAs for future research.
Discussion
In bacteria, sRNAs (~20 to 500 nucleotides) are a part of the regulatory network involved in diverse processes such as quorum sensing, carbon metabolism, stress response, and virulence. 67 Largely, sRNAs have been detected in the IGRs of the genome. However, recent studies reveal the synthesis of sRNAs from UTRs of genes and internal fragments of mRNAs, which is aided by alternate methods such as premature termination of mRNAs, Rnase E dependent mRNA decay, and internal transcription start site (iTSS). As a result of such diversity in the synthesis of these transcripts, the aforementioned methods can be inefficient in probing sRNAs across the genome. The establishment of sRNA-seq technology in the field of sRNA biology has assisted in understanding the contribution of sRNAs in the regulation of expression at multiple stages of growth. However, currently, available methods for the analysis of sRNA-seq data are not designed efficiently for the de novo identification of sRNAs in bacteria. Using POSIF, we identified 1120 sRNAs and 46 small proteins across multiple growth conditions in M. tuberculosis (see “Methods” section).
Conservation of the sRNA across closely related species signifies a potential role of these regions in the survival and growth of bacteria. Although much has been studied about the transcription factor/protein-mediated stress response in M. tuberculosis, sRNA-mediated stress response and its context-dependent expression are poorly understood. We studied the context-dependent expression of the identified sRNAs which revealed many sRNAs, including ncRv0188u and ncRv0038, which are expressed in a stress-dependent manner. On the other hand, sRNAs ncRv0146, ncRv0240uB, and ncRv2674i were expressed only in the rich medium growth. sRNAs distinctively expressed in all stress conditions, suggesting their importance in general stress response. The involvement of the putative targets of these sRNAs in the cellular and metabolic processes highlights the role of sRNAs in regulating these targets to optimize the growth of bacteria upon stress induction. We observed that some of these predicted sRNAs could be synthesized via alternate TSS and RNase E-mediated mRNA decay. Further studies on these putative sRNAs and small proteins will provide a detailed understanding of the relevance in M. tuberculosis stress adaptation.
In this study, we developed POSIF, a sRNA detection tool in bacteria. In contrast to other existing tools, POSIF is efficient in de novo identification of bacterial sRNAs using sRNA-seq data. POSIF operates without the need for dependencies and is not restricted to a specific operating system. Since POSIF is executed on the server and accessed via an API, it does not possess any significant client-side dependencies or computational resources. The User-Interface is friendly and explains all the details and has been provided with an example run case for bacterial sRNA prediction. Since POSIF is an expression data-based tool, data quality is important in determining its results. We elaborated on its utility and value in the case of the sRNA-seq data in M. tuberculosis. We hope that this robust feature of POSIF can efficiently enhance the strength of sRNA identification in bacterial genomics.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322241263674 – Supplemental material for Devising Isolation Forest-Based Method to Investigate the sRNAome of Mycobacterium tuberculosis Using sRNA-seq Data
Supplemental material, sj-docx-1-bbi-10.1177_11779322241263674 for Devising Isolation Forest-Based Method to Investigate the sRNAome of Mycobacterium tuberculosis Using sRNA-seq Data by Upasana Maity, Ritika Aggarwal, Rami Balasubramanian, Divya Lakshmi Venkatraman and Shubhada R Hegde in Bioinformatics and Biology Insights
Supplemental Material
sj-pdf-2-bbi-10.1177_11779322241263674 – Supplemental material for Devising Isolation Forest-Based Method to Investigate the sRNAome of Mycobacterium tuberculosis Using sRNA-seq Data
Supplemental material, sj-pdf-2-bbi-10.1177_11779322241263674 for Devising Isolation Forest-Based Method to Investigate the sRNAome of Mycobacterium tuberculosis Using sRNA-seq Data by Upasana Maity, Ritika Aggarwal, Rami Balasubramanian, Divya Lakshmi Venkatraman and Shubhada R Hegde in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-3-bbi-10.1177_11779322241263674 – Supplemental material for Devising Isolation Forest-Based Method to Investigate the sRNAome of Mycobacterium tuberculosis Using sRNA-seq Data
Supplemental material, sj-xlsx-3-bbi-10.1177_11779322241263674 for Devising Isolation Forest-Based Method to Investigate the sRNAome of Mycobacterium tuberculosis Using sRNA-seq Data by Upasana Maity, Ritika Aggarwal, Rami Balasubramanian, Divya Lakshmi Venkatraman and Shubhada R Hegde in Bioinformatics and Biology Insights
Footnotes
Author Contributions
Conceptualization: SRH. Formal analysis: UM, RA, RB, DLV, SRH. Data Curation: UM, RA, RB, DLV. Methodology: UM, RA, RB, DLV. Investigation: SRH. Writing – original draft: UM, RA, SRH. Writing – review and editing: UM, RA, SRH. All authors reviewed and approved the final version of the manuscript.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Department of Biotechnology, Ministry of Science and Technology, India grant—DBT-BUILDER to IBAB, Bengaluru, and Department of Electronics, IT, BT, and S&T, Government of Karnataka.
Data Availability Statement
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.