
Abstract
Coronaviral disease 2019 (COVID-19) is a recent pandemic disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Currently, there are still cases of COVID-19 around the world that can develop into persistent symptoms after discharge. The constellation of symptoms, termed long COVID, persists for months and can lead to various diseases such as lung inflammation and cardiovascular disease, which may lead to considerable financial burden and possible risk to human health. Moreover, the molecular mechanisms underlying the post-pandemic syndrome of COVID-19 remain unclear. In this study, we aimed to explore the molecular mechanism, disease association, and possible health risks in convalescent COVID-19 patients. Gene expression data from a human convalescent COVID-19 data set was compared with a data set from healthy normal individuals in order to identify differentially expressed genes (DEGs). To determine biological function and potential pathway alterations, the GO and KEGG databases were used to analyze the DEGs. Disease association, tissue, and organ-specific analyses were used to identify possible health effects. A total of 250 DEGs were identified between healthy and convalescent COVID-19 subjects. The biological function alterations identified revealed cytokine interactions and increased inflammation through NF-κB1, RELA, JUN, STAT3, and SP1. Interestingly, the most significant pathways were cytokine-cytokine receptor interaction, altered lipid metabolism, and atherosclerosis that play a crucial role in convalescent COVID-19. In addition, we also found pneumonitis, dermatitis, and autoimmune diseases. Based on our study, convalescent COVID-19 is associated with inflammation in a variety of organs that could lead to autoimmune and inflammatory diseases, as well as atherosclerosis. These findings are a first step toward fully exploring the disease mechanisms in depth to understand the relationship between post-COVID-19 infection and potential health risks. This is necessary for the development of appropriate strategies for the prevention and treatment of long COVID.
Introduction
The global pandemic coronaviral disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2, or SARS-CoV-2, is one of the most common transmissible RNA viruses, which continues impacting millions of patients. 1 Even though various types of vaccines and drugs against the virus have been developed, multiple viral mutants with increasing transmissibility and infectivity are emerging constantly. 2
In its most severe manifestation, SARS-CoV-2 infection leads to severe pneumonia symptoms and other consequences, such as pulmonary edema, acute respiratory distress syndrome, acute kidney damage, and multiple organ failure, that develop relatively quickly after viral infection. 3 Over 70% of COVID-19 patients with severe disease progress rapidly to acute respiratory distress syndrome, metabolic acidosis, and multiple organ failure. 4 The evidence suggests that aberrant activation of immunity, the elevation of plasma cytokines (tumor necrotic factor (TNF), IL-10, IL-6, and others), and cytokine storm are the major causes of multi-organ dysfunction in SARS-CoV-2 infected individuals. 5 Epidemiological studies indicate that acute and chronic kidney injury has a significant effect on patients’ prognoses. 6
Increasingly, medical research has shifted its focus to the genomic aspects of disease and global network analysis-based strategies which have allowed a deeper examination of disease pathogenesis and mechanisms. Innovative bioinformatics approaches have been employed to determine the association between gastrointestinal and cardiovascular diseases and COVID-19 infection. 7 In this regard, transcriptome analyses of non-coding and coding elements should be able to identify risk factors at the molecular level that can be used to identify individuals that are likely to develop severe COVID-19, thus allowing for the development of early intervention strategies and customized therapeutic options. 8 Gene expression profiling has been conducted in individuals infected with SARS-CoV-2 to characterize the association of certain gene expression patterns with the disease. 9 Due to the fact that functional relationships between gene products were not taken into consideration, associations were limited to the transcript level only. 10 Integrative network studies in medicine are important in order to understand the molecular mechanisms behind disease pathogenesis and to recognize interactions between biomolecules that play crucial roles in biological processes in both cells and tissues. 11 Recently, an integrative analysis was applied to identify molecular biomarker profiles for COVID-19 using transcriptome analyses in peripheral blood cells. 12
In this study, we aimed to identify the biological pathways and potential mechanisms involved in the disease process and their associations with specific affected organs using convalescent COVID-19 transcriptome profiles. The dataset of convalescent COVID-19 was selected for transcriptome analysis to identify the differentially expressed genes (DEGs). Differentially expressed genes were further analyzed for gene ontology, pathway enrichment, and protein network construction to identify biological alterations. Disease association and organ-specific analyses were performed to determine the health risk for COVID-19 post-pandemic syndrome and aid in the development of prevention and intervention strategies.
Materials and Methods
Data acquisition
The dataset GSE211378 investigated the cellular immune response in people who had recovered from SARS-CoV-2 infection. 13 The dataset consists of mRNA expression profiles that were generated using the GPL32571 nCounter Host Response Panel v1.0 platform. The dataset contains 304 whole blood samples from 264 COVID convalescent donors and 40 from healthy donors.
Identification of DEGs
GEO2R is a freely available interactive web tool (http://www.ncbi.nlm.nih.gov/geo/geo2r) comparing two groups of samples under the same conditions. 14 The dataset was used to find DEGs between healthy normal and COVID-19 convalescent individuals. The criteria were set at P < 0.05. The DEGs with logFC > 01 and logFC < 01 were regarded as upregulated genes and downregulated genes, respectively.
Gene ontology and enriched pathway analysis
DAVID Bioinformatics Resources 6.8 was utilized to distinguish and enrich the biological attributes, including molecular functions biological processes, cellular components, and enriched pathways of DEGs (https://david.ncifcrf.gov/). 15 Moreover, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and gene ontology (GO) were used for pathway enrichment analysis. The DEGs were used to identify the significant pathways. P < 0.05 was set as the cut-off criterion for significant enrichment.
Protein-protein network interaction and modular identification
The STRING database (Search Tool for the Retrieval of Interacting Genes https://string-db.org) is commonly performed to predict and construct protein-protein interaction (PPI) networks in bioinformatics. 16 We uploaded significant genes to the website and downloaded information for further analysis. Cytoscape (version 3.6.0), a freely available visualization software, was used to examine the relationship between DEGs. 17 Meanwhile, the MCODE plugin of Cytoscape was applied to screen central nodes in the PPI network by using the following criteria: degree cut-off = 2, node score cut-off = 0.2, k-core = 2, and maximum depth = 100.
Disease and tissue association analysis
To investigate disease and tissue-specific associated convalescent COVID-19, the DEGs were performed association and enrichment analysis using the DisGeNET database. 18 In addition, we also identified tissue-specific genes. The DEGs of convalescent COVID-19 subjects were applied to the Metascape database (https://metascape.org) and visualized by the “ggplot2” package. 19 Metascape is a freely available database of gene-disease and tissue-specific associations that contains one of the largest publicly available collections of genes and human disease-related variants. The cut-off criteria were set as follows: P-value ⩽ 0.05, enrichment ⩾ 1.5, and minimum count = 3.
Construction of PPI network and screening of hub genes
In our study, the PPI network of DEGs was constructed by using the STRING database, genes with a score of more than 0.4 were chosen to build the network and visualized by Cytoscape. In the PPI network, the betweenness centrality algorithm was used to find hub nodes. The betweenness centrality of each node was calculated by using the CytoHubba plugin. 20 In this study, the genes with the top 10 values were considered as hub genes.
Identification of transcription factor and microRNA regulations
To identify the enrichment of transcription factors (TFs), the DEGs were analyzed by using the Transcriptional Regulatory Relationships Unraveled by Sentence-based Text-mining (TRRUST) database. 21 The enriched TFs were plotted. Terms with a P-value < 0.01, a minimum count of 3, and an enrichment factor > 1.5 were screened out. The target genes of miRNAs were predicted by the online database miRDB based on thousands of miRNA-target interactions from high-throughput sequencing. 22 Tumor factor-miRNA regulations interaction network was predicted and constructed by using the NetworkAnalyst database.
Statistical analysis
Statistical analysis was performed by using the bioinformatic tools mentioned above. A P-value < 0.05 was considered statistically significant.
Results
Identification of DEGs
For the GSE211378 dataset, the gene expression data from healthy normal and convalescent COVID-19 subjects were normalized and analyzed for DEGs. Between the two groups, a total of 250 DEGs were identified consisting of 173 upregulated genes and 77 downregulated genes. A hierarchical clustering heatmap was constructed as shown in Figure 1A and B.

Differentially expressed genes (DEGs) identification among healthy normal and convalescent COVID-19 patients. (A) In a volcano plot of DEGs, red represents upregulated genes, and blue represents downregulated genes. (B) An overall clustering heat map of the DEGs. Red: upregulation; Blue: downregulation.
Biological and pathway enrichment
For GO enrichment analysis, the top three significant terms showed that response to cytokine, cytokine-mediated signaling pathway, and cellular response to cytokine stimulus were mainly involved in the biological process (BP). For the cellular compartment (CC), the GO analysis showed that the external side of the plasma membrane, side of membrane, and cell surface were mostly enriched. In molecular function (MF), cytokine receptor binding, cytokine receptor activity, and signaling receptor binding were significantly enriched. The GO dot plot is shown in Figure 2A to C.
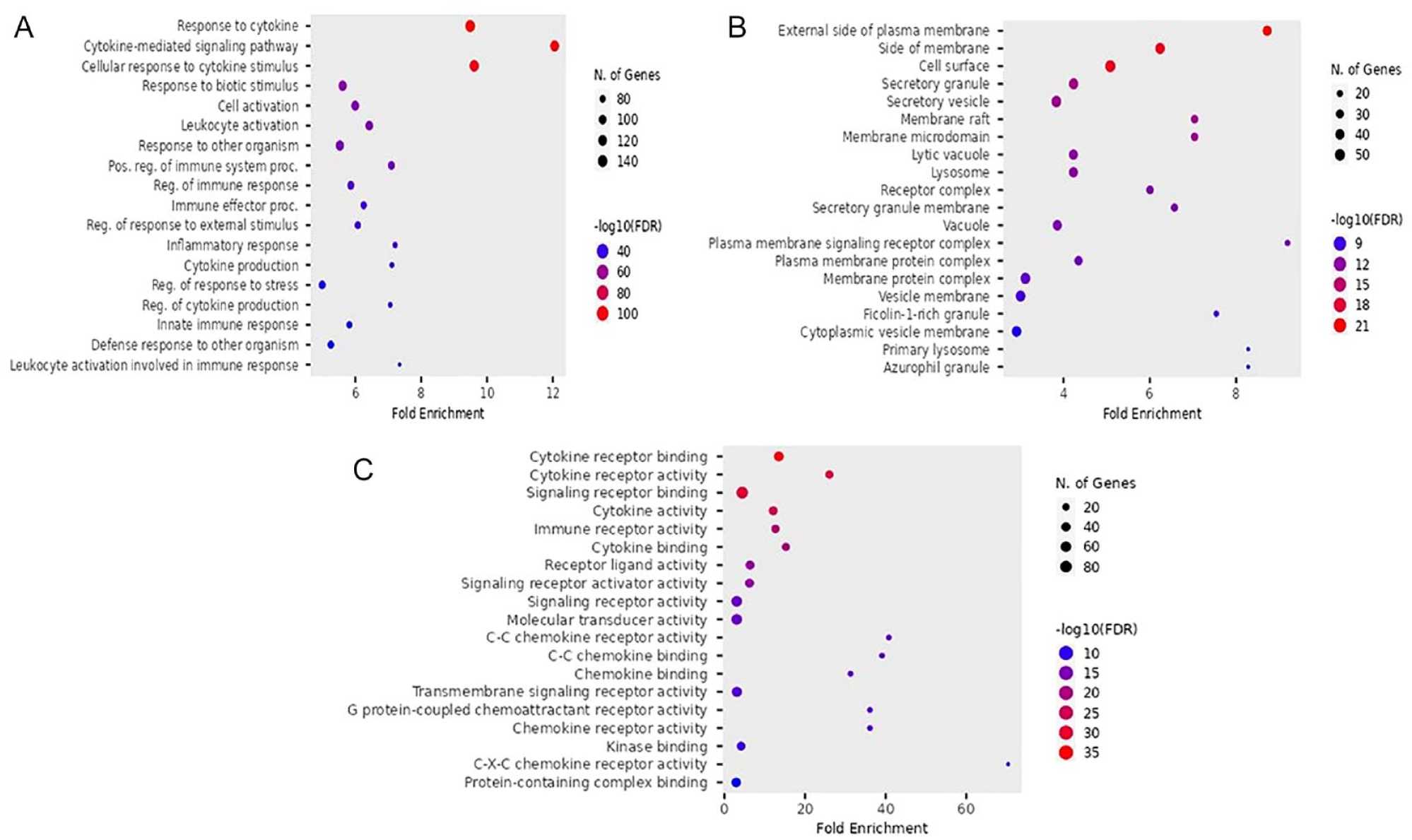
Gene ontology function enrichment analysis. (A) Biological process (BP). (B) Cellular component (CC). (C) Molecular function (MF). FDR, false discovery rate.
In terms of KEGG pathway enrichment analysis and KEGG networking, cytokine-cytokine receptor interaction and lipid atherosclerosis were mainly enriched in both pathway enrichment and their network as shown in Figure 3A and B. The results indicated that the cytokine-cytokine receptor interaction and lipid and atherosclerosis may associate with convalescence COVID-19 as demonstrated in Figures 4 and 5.

(A) Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis. (B) The network of pathway enrichment. JAK indicates Jun amino-terminal kinase; STAT, signal transduction and activator of transcription; IL, interleukin; NOD, nucleotide-binding oligomerization domain-like receptors; NF, nuclear factor; FDR, false discovery rate.

The KEGG pathway of cytokine-cytokine receptor interaction. KEGG indicates Kyoto Encyclopedia of Genes and Genomes; TNF, tumor necrotic factor; TGF, transforming growth factor.
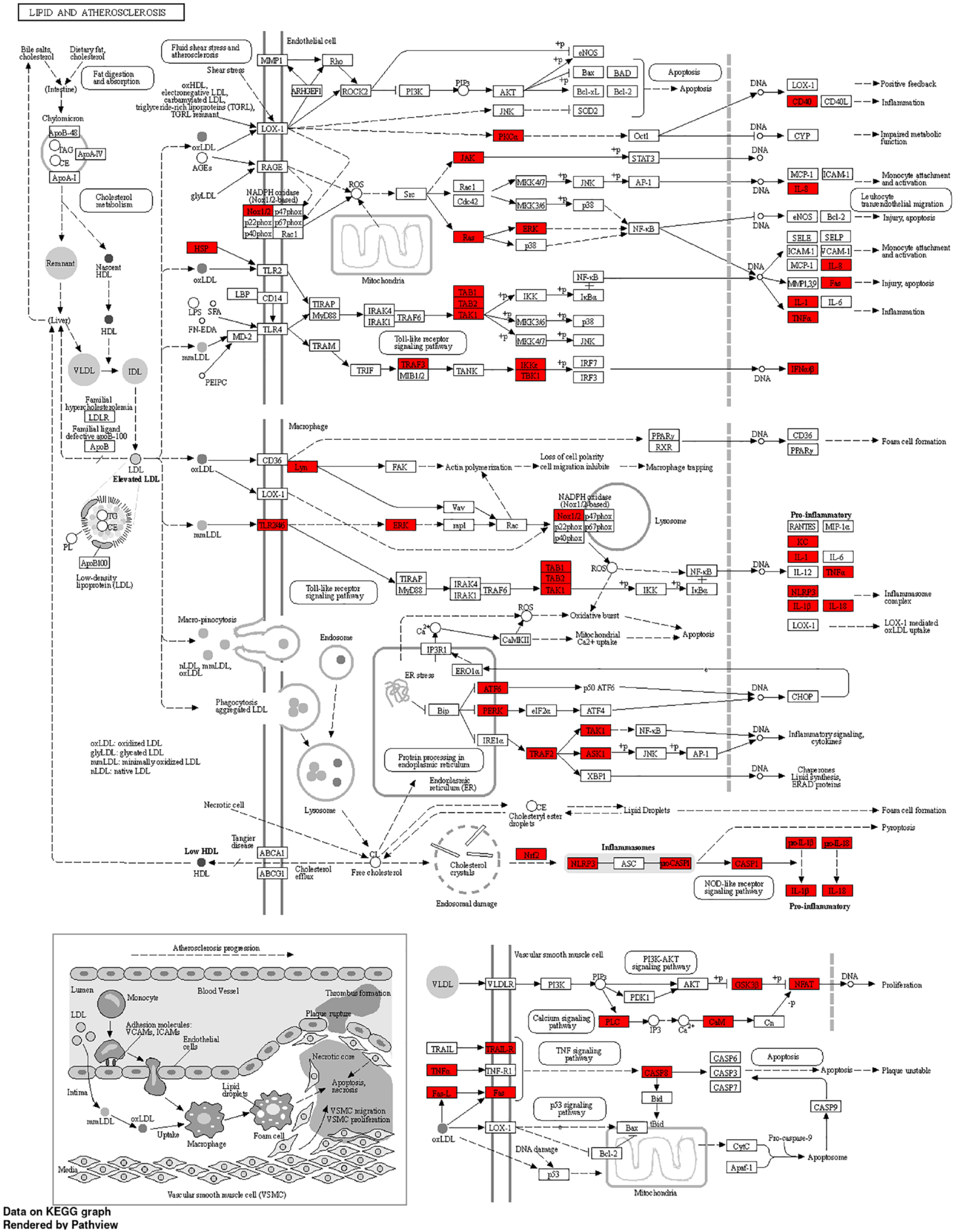
The KEGG pathway of lipid and regulatory interactions leading to atherosclerosis. ERK indicates extracellular signal-regulated kinase; JNK, Jun amino-terminal kinase; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Protein networking and modular analysis
Using the STRING database and Cytoscape, the PPI network of the DEGs consisted of 246 nodes and 3358 edges, accompanied by an average node degree of 27.3 and a local clustering coefficient value of 0.539. MCODE plugin was used to construct the functional clusters; 2 clusters had an MCODE score > 5. A total of 9 MCODEs were identified as in Figure 6. The terms of the MCODEs are listed in Table 1.

(A) All protein-protein interactions (PPI) among all differentially expressed genes. (B) Gene ontology enrichment analysis was applied to MCODE module analysis for sub-network categorization and each MCODE network is assigned a unique color.
The results of MCODE enrichment analysis.
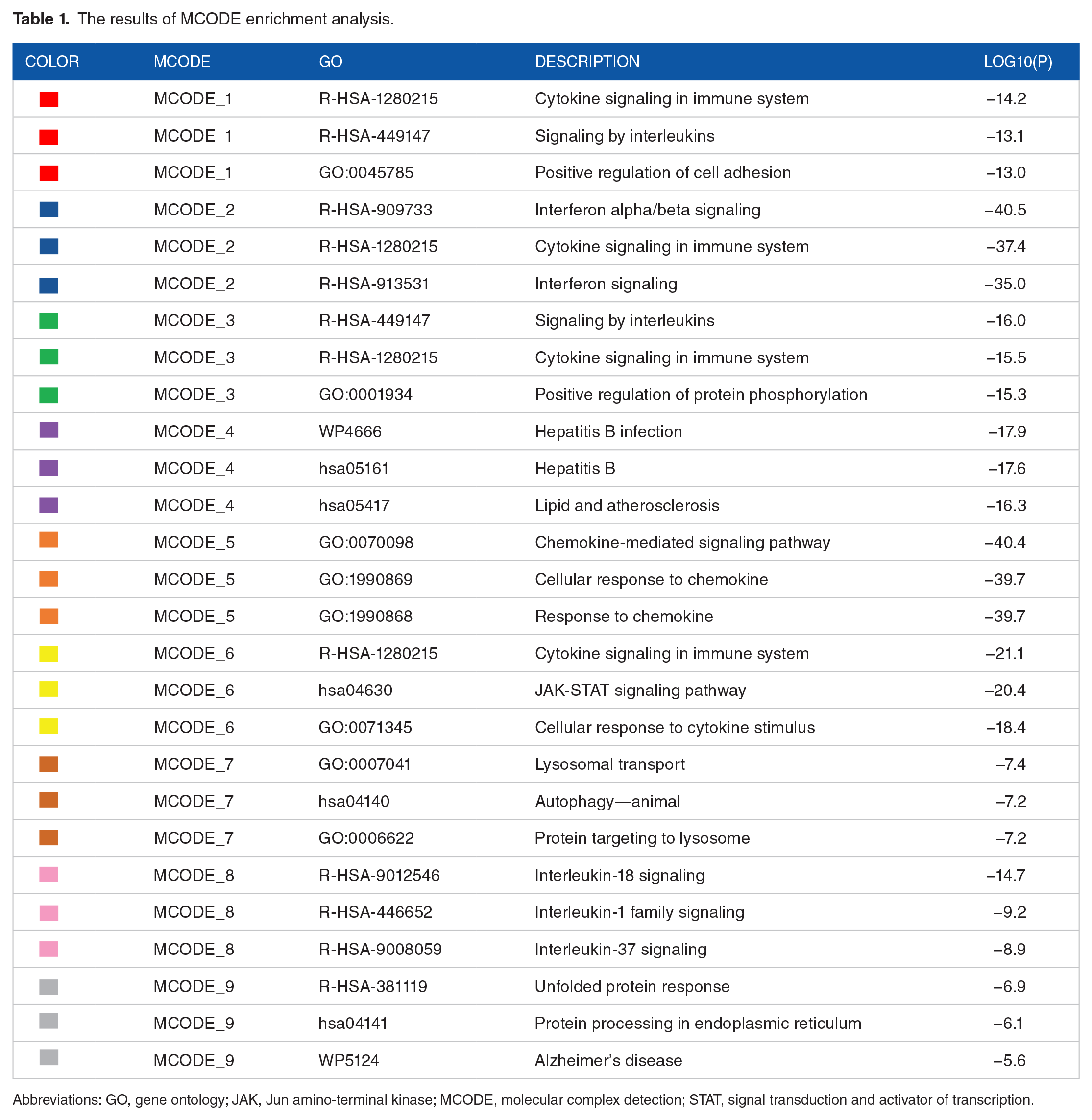
Abbreviations: GO, gene ontology; JAK, Jun amino-terminal kinase; MCODE, molecular complex detection; STAT, signal transduction and activator of transcription.
In order to further focus on the relationships among the terms, a subset of terms was selected and rendered as a network plot, where the terms with a similarity > 0.3 are connected by edges. The terms with the best P-values were selected from each of the 20 clusters, with the constraint that there were no more than 15 terms per cluster and no more than 250 terms in total. The network was visualized using Cytoscape software, where each node represents an enriched term and was colored first by its cluster ID and then by its P-value, as shown in Figure 7. The pathway and biological process enrichment analysis was carried out with the following ontology sources: GO Biological Processes, Reactome Gene Sets, KEGG Pathway, Canonical Pathways, WikiPathways, PANTHER Pathway, and CORUM. Terms with a P-value < 0.01, a minimum count of 3, and an enrichment factor > 1.5.

The network of enriched terms was labeled by P-value in which the terms containing more differentially expressed genes tend to have a more significant P-value. JAK indicates Jun amino-terminal kinase; STAT, signal transduction and activator of transcription.
PPI network construction and hub genes selection
Based on the PPI network, the DEGs were obtained and the PPI was visualized by the Cytoscape software. The top 10 hub genes were screened out by centrality betweenness score including TNF (score: 4693), RIGI (score: 3887), MAPK1 (score: 3818), JAK2 (3365), PTPN6 (score: 3065), LYN (score: 2610), HSP90AB1 (score: 2463), PRKCA (score: 2316), CXCR4 (score: 2023), and LRRK2 (score: 1957), as shown in Figure 8.
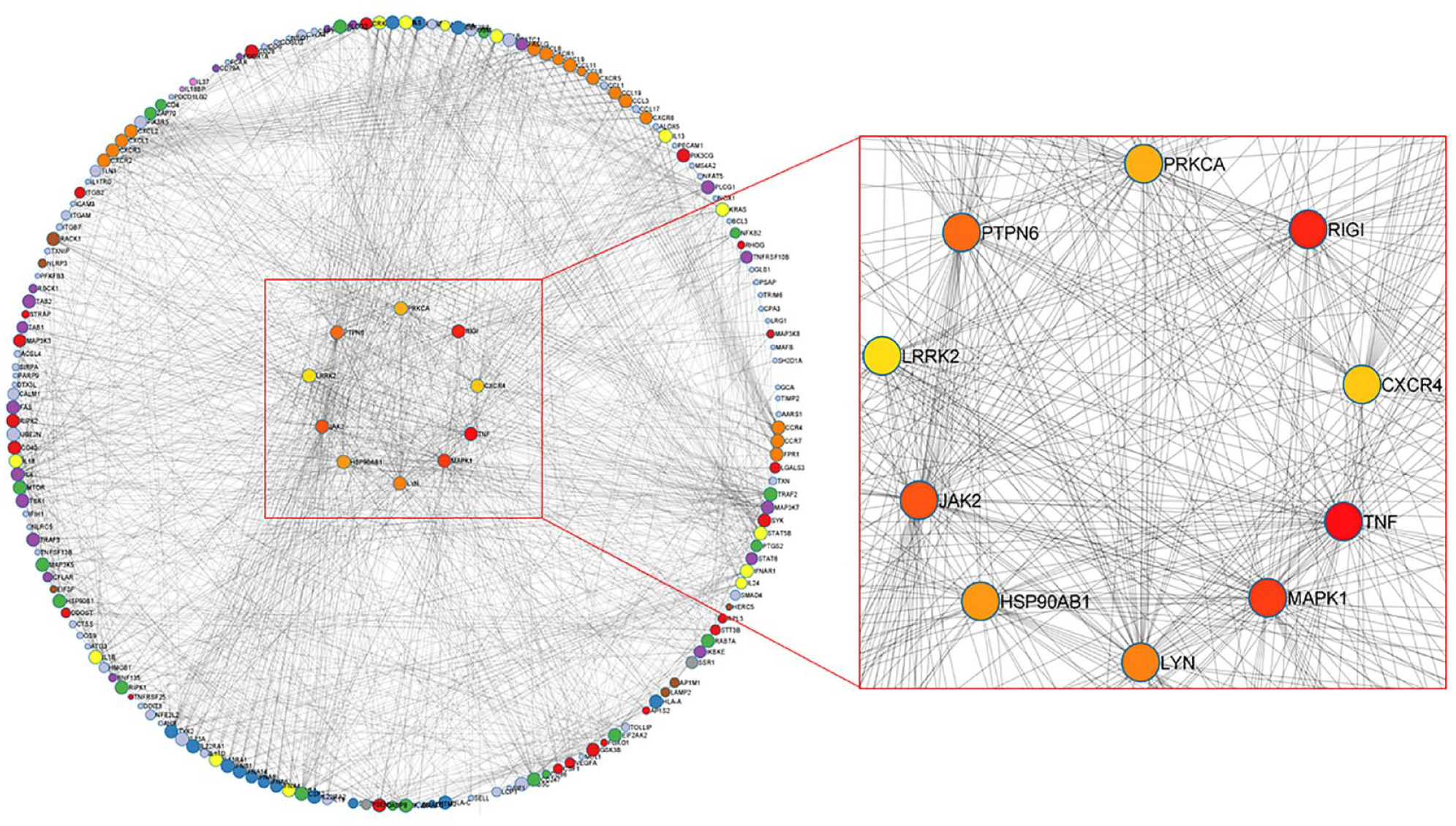
Protein-protein interaction (PPI) network and the top 10 hub genes.
Disease association and tissue-specific analysis
Diseases associated with COVID-19 convalescence were also investigated. The disease enrichment analysis demonstrated that pneumonitis, infection, and dermatitis were associated with convalescent COVID-19, as shown in Figure 9. The PaGenBase database was used to identify convalescent COVID-19-associated tissue-specific terms. The tissue-specific analysis showed that the DEGs were mainly enriched in the spleen, blood, and lung, respectively, as shown in Figure 10.

Bar graph of enriched terms in disease association analysis of the differentially expressed genes.

Bar graph of enriched terms from tissue/organ-specific analysis (coloring by P values).
Identification of TF and miRNA regulations
Transcription factor identification analyses of the DEGs using TRRUST database revealed: NF-κB1, RELA, JUN, STAT3, and SP1, to be the five most enriched TFs regulating the convalescence of COVID-19, as shown in Figure 11A.

(A) Bar chart of transcription factors (TFs) regulation prediction determined using Metascape (colored by P value). (B) The miRNA-TFs interaction network.
To further investigate the potential molecular mechanisms of convalescence of COVID-19, we constructed a TF-miRNA interaction network. MYC was the most interaction with mA including hsa-mir-98-5p, hsa-let-7d-5p, hsa-mir-20a-5p, hsa-mir-24-3p, hsa-mir-155-5p, hsa-mir-16-5p, hsa-mir-17-5p, hsa-mir-34a-5p, hsa-mir-26b-5p, hsa-mir-1-3p, hsa-let-7a-5p, hsa-let-7b-5p. hsa-mir-335-5p, hsa-mir-10b-5p, hsa-mir-103a-3p and hsa-let-7e-5p, as shown in Figure 11B.
Discussion
Coronavirus disease 2019, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has emerged as a global public health crisis. 23 A previous report demonstrating that the survivors of SARS-CoV-1 and MERS-CoV infection showed that persistent symptoms or underlying organ injury, such as acute kidney injury, acute liver injury, acute respiratory distress syndrome, and acute myocardial injury, were sometimes present despite recovery from the initial infection. This matched with the multi-organ injuries that are observed in some COVID-19 patients following discharge.24,25 Therefore, in the present study, we aimed to investigate the potential molecular mechanisms of pathogenesis, associated diseases, and tissue/organ-specific events that may have an effect during COVID-19 convalescence using transcriptome bioinformatics and network analysis.
Mechanistically, angiotensin-converting enzyme 2 (ACE2) mediates SARS-CoV-2 infection of endothelial cells (ECs) causing endothelial dysfunction and dysregulation of the renin-angiotensin system. 26 The dysfunction of ECs is an early step in the development of atherosclerosis that precedes clinical symptoms and has a prognostic value for future cardiovascular events. 27 Although there is no result showing the relationship between ACE2 expression and atherosclerosis mechanisms in the present study, it deserves further investigation.
Innate and adaptive immune responses play an important role in defense mechanisms against viral infection and during disease progression. Natural killer (NK) cells provide primary control during acute viral infection, but cytotoxic CD8 T lymphocytes (CTLs) are crucial for long-term surveillance. 28 However, increased elicitation of immune responses following infection has been associated with the excessive production of proinflammatory cytokines and ubiquitous tissue damage. 28 Preliminary study has shown that SARS-CoV-2 infection triggers a cytokine storm and increases various cytokine levels. 29 Previous studies have shown that high levels of chemokines and cytokines mediate tissue inflammation during infections that can lead to cellular damage. 30 From our analysis, cytokines stimulation and cytokines interaction were mostly enriched and MCODE module analysis showed a variety of cytokines, such as immune, IL1b, and IL-37, play an important role in immuno-modulation and inflammation activation in convalescent COVID-19. IL-18 and IL-1β are crucial proinflammatory cytokines that play important roles in innate and adaptive immune responses. 31 IL-1β also increases the production of IL-6 and TNF, while its production is stimulated by both cytokines providing an integrated amplification of the inflammatory response. 32 Of note, IL-18 binding protein (IL-18BP) in high levels also binds IL-37, preventing IL-37 to suppress inflammation. 33 IL-37 is an anti-inflammatory cytokine that represses both the innate and adaptive immune responses. 34 Accordingly, the administration of recombinant IL-18 could induce the sequestration of IL-37, thus possibly inducing inflammation.
For PPI network and hub gene selection, TNF was identified as the top hub gene that has a crucial role in convalescent COVID-19. Tumor necrosis factor is one of the most critical proinflammatory cytokines produced by several immune cells such as natural killer (NK), monocytes, macrophages, neutrophils, and T-cells. 35 Tumor necrosis factor expressed mainly in the acute phase of infections and acted as a key regulator in the immune response. 36 Tumor necrosis factor triggers complex signaling where a low level benefits the health, but high levels can promote organ damage. 37 In the case of COVID-19, TNF is commonly upregulated in acute lung injury, triggers (cytokine release syndrome) CRS, and facilitates SARS-CoV-2 interaction with ACE2. Inhibition of TNF may serve as an effective therapeutic strategy for attenuating disease progression in COVID-19 infection. 38
According to the present study, TF analysis indicated that NF-κB, RELA, JUN STAT3, and SP1 may play important roles during COVID-19 convalescence. COVID-19 disease activation of the transcription factor, NF-kappa B (NF-κB), in cells such as macrophages in the cardiovascular system, gastrointestinal system, central nervous system, lung, liver, and kidney, leads to the production of IL-1, IL-2, IL-6, IL-12, GM-CSF, LT-β, LT-α, TNF-α, as well as various chemokines. 39 Coronavirus infection has been shown to involve extracellular signal-regulated kinases (ERK1/2, MAPKs, the Jun amino-terminal kinases (JNK), and P38 pathways for viral infection and disease pathogenesis.40,41 The Janus Kinase (JAK) and signal transduction and activator of transcription factor 3 (STAT3) pathway are required for activation of the NF-κB pathway and IL-6, which are inflammation stimulators. 42 Interleukin-6 binds to its receptor, activates JAK-STAT signal transduction, and allows phosphorylation of STAT3, which stimulates its translocation to the nucleus to repress interferon (IFN)-γ production and induces cytokine release syndrome. 43 Our further analysis showed that MYC had the highest interaction with miRNA and was regulated by multiple miRNAs. Previous study revealed that c-Myc plays a key role in inflammation regulation that was regulated by a variety of miRNAs such as miRNA-103 and miRNA-145.44,45 In addition, MYC is one of the most commonly overexpressed oncogenes in cancer and one of the most robust and significant prognostic markers for B-cell lymphomas. MYC dysregulation has been implicated in the aggressive transformation of B-cell lymphomas. 46 Although earlier studies of miRNA suppressing gene expression have been numerously reported, however, for MYC, our results showed multiple interactions with miRNA, thus the lacking of reported study of miRNA-MYC regulation should be investigated further. At present, the molecular mechanism in convalescent COVID-19 patients remains unclear and this finding demonstrates that inflammatory cytokines still appear although without COVID-19 detection.
Coronaviral disease 2019 is mostly a respiratory infection. However, both autopsy findings and clinical observations have detected vascular damage and thrombotic complications in various organs. 47 In this study, KEGG pathway analysis found that lipids and atherosclerosis play a crucial role during COVID-19 convalescence. Atherosclerosis is a chronic inflammatory disease of the arterial intima, which is lipid-driven maladaptation. It is characterized by the dysfunction of lipid homeostasis and signal transduction that regulates inflammation. 48 In the event of COVID-19, an alteration in lipid metabolism was observed, which was mainly caused by HDL impairment. 49 According to the present study, we also found that a variety of cytokine-cytokine interactions were enriched in KEGG pathway analysis. The activation of immune cells leads to the development of uncontrolled inflammation and dysregulation of the immune system namely cytokine storm, as well as the accumulation of eicosanoids, such as lipoxin A4 (LXA4), thromboxane B2 (TXB2), prostaglandin E2 (PGE2), and leukotriene B4 (LTB4). 50 The uncontrolled inflammation leads to the impairment of HDL lipoprotein function through a reduction in the levels of apolipoprotein AI (ApoA-I), apolipoprotein E (ApoE), and increased serum amyloid A (SAA).51,52 These alterations reduce the antioxidant, anti-inflammatory, and immunomodulatory properties of HDL lipoproteins. 53 The effects of the interaction of oxLDL and LOX-1 (accumulation of intracellular oxLDL) stimulate the progression of atherosclerosis. 54 The use of cholesterol-lowering drugs may reduce the risk of severity of COVID-19 and prevent the risk of death in these patients and statin drugs may be used as a preventive drug to treat COVID-19 convalescence-related health effects.
While the adverse effect of COVID-19 infection on the respiratory system is well established, its impact on the liver and spleen has not been investigated much. Our analysis showed that the spleen and blood are the tissues that show the highest specific association with COVID-19 convalescence. The spleen plays an essential role in the regulation of the immune system, modulating the B and T cell responses to the antigenic targets in the blood system, and coronaviruses have a high tropism for the spleen. 55 Autopsy observations have revealed interesting anatomical changes in the spleen during coronavirus infection, including a reduction in the splenic cellular composition, and depletion of B and T lymphocytes. 56 The impairment of the spleen in COVID-19 patients has been poorly described. It is thought that impairment may be driven by a variety of mechanisms, including cytokine-mediated immune pathogenesis, lymphocyte apoptosis, microvascular dysfunction, and direct organ attack by the virus. 57 Indeed, further studies are needed to clarify the health impacts of SARS-CoV-2 in patients with spleen dysfunction. Further studies will lead to a better understanding of the health impact in SARS-CoV-2-infected patients and aid in the development of therapeutic approaches, especially for those with pre-existing spleen impairment, who seem to be at higher risk of a worse outcome. However, there are certain limitations to this study. One of the limitations of our study is the lack of complete health status of convalescent COVID-19 and the heterogeneity of individual patients because there are multiple factors causing atherosclerosis such as smoking, high cholesterol, and genetic factors. For sample selection, the conclusion is based on one dataset collected from a public database. Enough patient samples should be employed in the bioinformatics analysis to ensure more accurate results.
Conclusion
In this study, we explored the potential for new insights into the mechanisms underlying diseases related to COVID-19 convalescence in whole blood samples by using transcriptome bioinformatics analyses. Differentially expressed genes identified in COVID-19 convalescent patients were used for the investigation of GO, pathways, PPI networks, disease association, and tissue-specific analysis. We found that cytokine-mediated signaling is involved in biological alteration through GO analysis. Interestingly, pathway enrichment also showed the term lipid and atherosclerosis associated with convalescent COVID-19. However, modular analysis from protein networks points to a role for inflammation and stimulation of cytokines through the regulation of transcription factors such as NF-κB, RELA, JUN, STAT3, and SP1. In addition, convalescent COVID-19-associated disease analysis indicated the presence of inflammation in various organs, including the lungs, skin, and immune system, which could increase the risk of autoimmune disease development. These findings contribute novel insight into the mechanisms of convalescent COVID-19 and the possible health effects in post-COVID-19 syndrome that could lead to the development of prevention and intervention strategies in the future.
Footnotes
Acknowledgements
We sincerely thank Dr. James M. Dubbs Ph.D., senior research scientist at the Chulabhorn Research Institute (CRI), for proofreading the manuscript.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Chulabhorn Research Institute (CRI) Grant number: 4274379.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SC and JS designed and provided a conceptual framework for this study. SC, WN and TS performed data collection, data interpretation and data analysis. SC performed bioinformatics analysis and contributed to writing the first draft. All authors revised and approved the final version of manuscript.