
Abstract
The Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Omicron variant and its subvariants have a unique set of mutations. Two of those mutations (N679 K and P681 H) reside close to the S1 /S2 furin cleavage site (FCS; 685-686). When these mutations reside together, they exert less-efficient membrane fusion than wild type and most other variants of concern such as the Delta variant. Here, we in silico targeted these mutations to find out which of the amino acids and interactions change plays the key role in fusion. To comprehend the epistatic effect of N679 K and P681 H mutations on the spike protein, we in silico constructed three types of spike protein sequences by changing the respective amino acids on 679 and 681 positions (P681 H, N679 K, K679 N-H681 P variants). We then analyzed the binding affinity of furin and spike (Furin-Wild, Furin-Omicron, Furin-P681 H, Furin-N679 K, and Furin-K679 N/H681 P) complexes. Omicron and P681 H variants showed a similar higher binding energy trend compared to the wild type and N679 K. The variation in hydrogen, hydrophobic, and salt bridge bonds between spike protein and furin provided an explanation for the observed low fusogenicity of Omicron. The fate of the epistasis in furin binding and possible cleavage depends on the efficient interaction between FCS in spike and furin catalytic triad, and in addition, the loss of the hydrogen bond between Arg 681 (spike) and Asn 295 (furin) along with inhibitor-like ineffective higher affinity plays an important role in the enzymatic activity.
Introduction
The etiologic agent of the COVID-19 pandemic, the SARS-CoV-2 has evolved rapidly, producing new variants frequently. 1 The Omicron variant or B.1.1.529 pango lineage of SARS-CoV-2 is of great concern among them because it contains a large number of mutations relative to the original strain, including three regions of the viral surface spike protein that are extremely crucial for infection and escaping immunity (the receptor-binding domain, the furin cleavage domain [FCD], and the N-terminal domain).2,3 The fitness of a particular variant of virus usually depends on its evolutionary advantageous mutations playing roles in higher binding affinity to receptor, rapid replication, and fusogenicity of the virus. In case of SARS-CoV-2, the FCD appears to exert a strict influence and regulation over efficient membrane fusion and transmission.4-6
In the Omicron variant, a particular cluster of mutations (H655Y, N679 K, P681 H) are present at the FCD. These mutated residues positioned just before the S1 S2 cleavage site at 685-686. 7 The furin-mediated cleavage is the slowest and weakest in this variant even though it contains mutation at the 681 position. 8 Notably, the P681R mutation in the Delta variant gives strong fusogenicity and thus pathogenicity. 9 Then, why the Omicron variant has less and slower cleavage ability especially when it has multiple mutations near the cutting site, including P681 H? The question is important since this property is linked to reduced efficiency of spike cleavage by host proteases, which causes less-severe infection and disease in the host.
In this study, we target two mutations at FCD (N679 K and P681 H) that are closer to the furin cutting site to find out the molecular mechanism of previously experimentally proved epistatic (negative) effect of these mutations. We also explain the evolutionary road map of the selective pressure exerted on spike protein mutants while generating the final variants with both mutations. Our study depends on bioinformatics analysis based on molecular docking and dynamics. To understand why the Omicron variant has low fusogenicity, the molecular interactions need to be analyzed and discussed even though wet lab experiments were done showing fusogenic activity. Overall, this in silico approach unveils the mechanistic way of the spike-furin interaction in the Omicron variant.
Materials and Methods
Sequence retrieval and processing
The amino acid sequences of the spike protein wild type (accession no. NC_045512.2) and Omicron variant (accession no. OX315675.1) of SARS-CoV-2 were retrieved from the National Center for Biotechnology Information. 10 In order to comprehend the epistatic effect of N679 K and P681 H mutations on the spike-furin binding and interaction, we manually constructed spike protein sequences by changing the respective amino acids on 679 and 681 positions using MEGA version 7.0. 11 Finally, after a different combination, five types of target sequences of spike protein for this study were finally selected considering the constructive mutations as shown in Table 1 and Supplementary Figure 1. The reason for generating different spike proteins and evolutionary perspective is explained in Figure 1.
Mutations in the predicted computational constructs of the spike protein of SARS-CoV-2.

SARS-CoV-2: severe acute respiratory syndrome coronavirus 2.
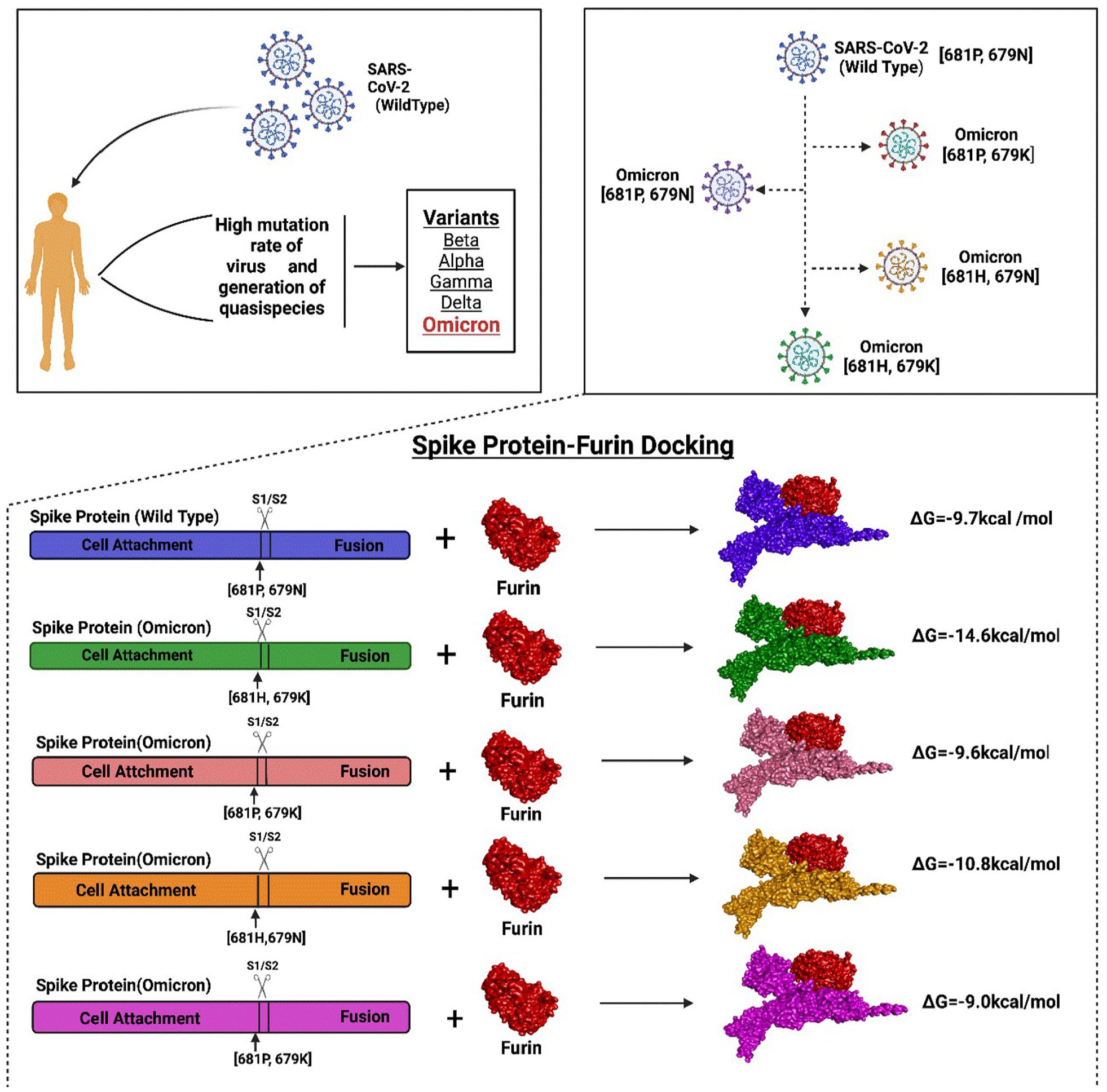
Graphical abstract of the possible quasi-species of Omicron variants and the interaction of their spike protein with furin.
Tertiary structure prediction and quality assessment
The tertiary structures of the spike protein for the constructed target sequences selected for the study were predicted through the SWISS-MODEL homology modeling webtool 12 using the template 7n1u.1. The predicted structures were further refined through the GalaxyRefine 13 server. After analyzing all the potential structures generated after refinement, arguably the one with the best quality and performance was selected. Evaluation of the refined 3D structures was done by PROCHECK, 14 ERRAT, 15 and Verify3D 16 modules of the SAVESv6.0 server (https://saves.mbi.ucla.edu/). The ExPASy server (https://www.expasy.org/) of the Swiss Institute of Bioinformatics incorporates different bioinformatics tools. Between these resources, the SWISS-MODEL Structure Assessment tool 12 and QMEAN tool 17 were collaboratively used to estimate the QMEAN Z score and global quality of the model.
Docking analysis
In order to find out the relative binding affinity and interaction of the spike protein of SARS-CoV-2 with furin, protein-protein docking analysis was performed using HADDOCK v2.4 tool, 18 PRODIGY, 19 and Schrodinger’s Maestro. The residue level protein-protein interaction was visualized through BIOVIA Discovery Studio 4.5 Software 20 and LigPlot + (v2.2). 21 The H-bonds and hydrophobic interactions were depicted using LigPlot + (v2.2), which is entailed with an in-built algorithm to calculate the bond distances. The salt bridges’ conventional and charged interactions were predicted, visualized, and calculated through BIOVIA Discovery Studio 4.5 software.
The proteins were also prepared with the Protein Preparation Wizard of the Schrodinger 2020-3 release 22 using default settings. The hydrogen bonds were optimized and a final restrained minimization of the system was carried out under the OPLS3e force field with a full optimization for hydrogen atoms and a 0.30 Å maximum RMSD (Root-Mean-Square Deviation) of the heavy atoms from the initial position. The protein-protein docking was performed using the rigid body docking program PIPER 23 module of Schrodinger 2020-3, which evaluates energy functions in a discretized 6D space of mutual orientations using FFT (Fast Fourier Transform). The SARS-CoV-2 spike protein was selected as the receptor and furin as the ligand. The output complexes were thoroughly checked for binding at the active site, with consideration given to PIPER Pose Energy and cluster size. The complexes were ranked based on cluster size and the pose energy function, which describes receptor-ligand interactions and is efficiently calculated using Fast Fourier Transforms.
Molecular dynamics study
The molecular dynamics simulations study was performed in YASARA Dynamics software 24 package where AMBER14 force field was applied. 25 The simulated complexes were initially optimized, and hydrogen bond networks were oriented. The TIP3 P water solvation’s model was used with periodic boundary conditions.26,27 The physiological conditions of the simulations cell were set as 298 K, pH 7.4%, and 0.9% NaCl. 28 The simulated complexes were initially energy minimized with the steepest gradient approaches using a simulating annealing method (5000 cycles). The time step of the simulations was set as 2.0 fs. The long-range electrostatic interactions were calculated by the particle mesh Ewald methods by a cutoff radius of 8.0Å.29,30 The simulations’ trajectories were saved after every 100 ps and extended for 30 ns time.
Results
Structural validation, docking analysis, and molecular dynamics simulation
The tertiary structures predicted through homology modeling showed higher confidence of prediction after refinement through energy minimization. The Ramachandran plots and Z scores indicated the structures to be valid and stable.
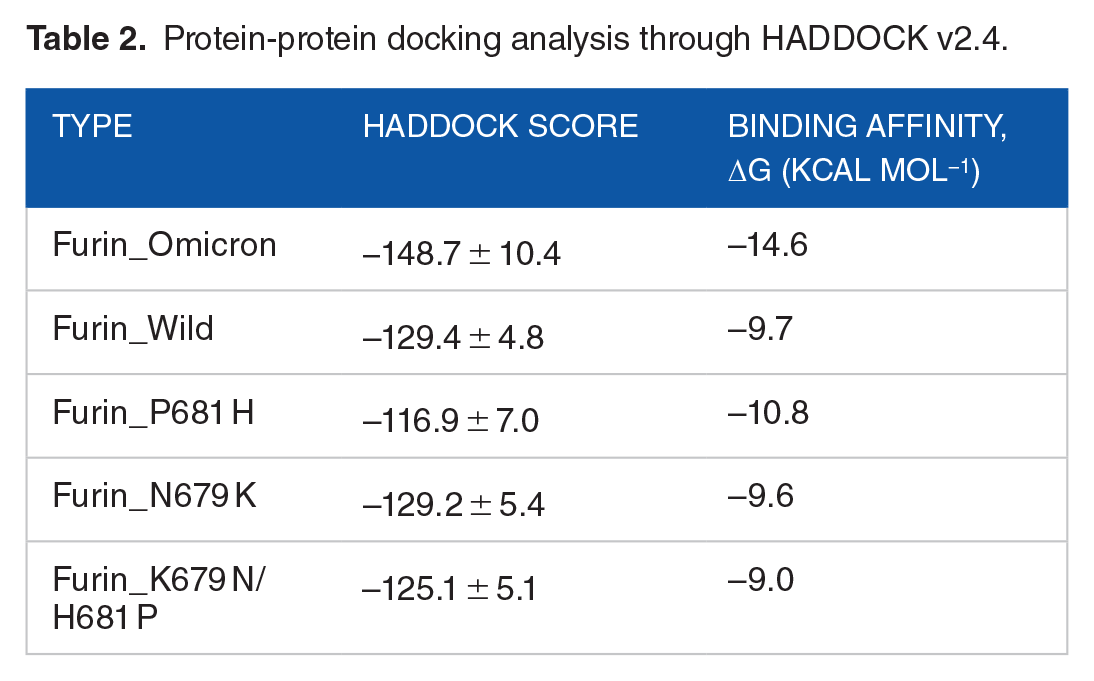
After the protein-protein docking analysis in HADDOCK v2.4, the cluster having structures with the lowest Z score was selected for further study, which ensures the selection of the best docking result with the highest confidence limit. The docking analysis of spike protein with furin revealed that the binding affinity of the spike protein of Omicron is higher than that of the wild type. Besides, the spike protein of Omicron variant with the P681 H mutation showed relatively higher binding affinity than the one with N679 K mutation. The docking results obtained from HADDOCK v2.4 and PRODIGY are summarized in Table 2. Furthermore, the protein-protein docking analysis carried out with a different algorithm using the PIPER module of Schrodinger 2020-3 software also manifested quite similar results obtained from HADDOCK. Here, mutation at the 681 position reveals the lowest pose energy and indicates better binding than the others. The wild one exhibits comparatively weaker binding than the others (Table 3). The docking was done by the blind docking method, so the docked complexes were checked for binding at the active site and taken into consideration. The output complexes are ranked by the cluster size, and the pose energy function describes the receptor-ligand interactions which are defined on this cluster and efficiently calculated using Fast Fourier Transforms. Finally, the interactions between furin and spike are shown in the Figures 2 and 3, and the results are combined in Table 4.
Protein-protein docking analysis through HADDOCK v2.4.
Protein-protein docking analysis through Schrodinger software.


The hydrogen (green lines) and hydrophobic (red Lines) bonds between the furin and different spike variants. Here chain A = spike protein and chain B = furin. (A) Wild type. (B) Omicron. N679 K, P681 H. (C) 679 K variant, N679 K, H681 P. (D) 681 H variant, P681 H, K679 N. (E) Delta variant, P681R.

Interaction of different variants of SARS-CoV 2 spike protein cleavage site with furin protease. (A) Wild, (B) Omicron, (C) N679 K variant, and (D) P681 H variant.
Specific interactions between spike furin cleavage site and furin’s active region.

Chain A = spike protein, Chain B = furin.
The molecular dynamics simulations study was conducted to analyze the stable nature of the protein complexes. The root mean square deviations of the C-alpha of the protein were also explored. Figure 4 demonstrated that all complexes, including N679 K + P681 H, N679 K, P681 H, Omicron, and wild, had an initial upper trend at the beginning of simulations, which indicates the complexes’ flexibility. Therefore, the complexes reached steady state after 15 ns and maintained the integrity till the rest of the simulation periods. The overall RMSD trend did not exceed 2.5Å, which defines the complexes’ stable nature.
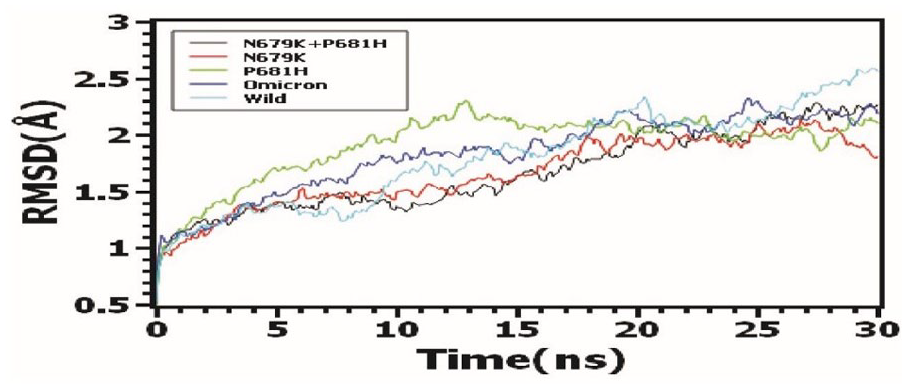
Molecular dynamics (MD) simulation of the protein complexes for the stability investigation through RMSD calculation.
Discussion
The Fusion property of the SARS-CoV-2 is significant because of its link to cell entry and thus pathogenesis and disease severity.1,5 The first cleavage event mediated by furin occurring at the S1/S2 cleavage site enables further processing of the spike protein by other protease/s at the S2′ site.31-33 Therefore, effective binding of the furin with spike protein’s multi-basic (R-R-A-R) cleavage site is extremely important. Particular mutation(s) in spike protein can affect this fusogenicity through modulating binding with and cleavage capacity of the host proteases. Our study is about understanding the epistatic effect of two particular mutations (N679 K and P681 H) of the Omicron variant, which are adjacent and immediately upstream to the furin cleavage site (FCS) of the spike protein (685-86 position). The other mutation near the FCD such as H655Y critically governs the low fusogenicity of Omicron through dictating the enhanced endosome entry pathway utilization, which is not linked to furin-based cleavage. 34 In addition, while the evolution of the Omicron variant was occurring in a population, there could be three quasi-species circulating, which finally produced the final variant with both N679 K and P681 H mutations (Figure 1). Since the study was related to the interaction between furin and SARS-CoV-2 spike protein, the tertiary structures of the spike protein targeting the respective mutations of those quasi-species were modeled and further subjected to docking analysis in order to find out the binding affinity of furin with spike protein as well as the efficiency of cleavage. The highest binding affinity was measured for the Omicron variant or P681 H mutant (Tables 2 and 3). The molecular dynamics study showed that all the complexes had lower RMSD points, which indicated the stability of the complexes at simulation conditions (Figure 4).35,36 Importantly, the P681 H mutant showed higher fusion capacity in vitro. 37 But, the Omicron has slower and less fusogenicity even though it showed strong binding with furin in our analysis and previous studies.38,39 The reasons can be two-fold: Higher binding can actually make it difficult for furin to detach after cleavage, and chaning of amino acid R-groups may affect appropriate binding with furin to cut the specific site easily.
This study demonstrates that P681 H mutation in the spike protein of SARS-COV-2 provides the major contribution to the stability of the furin-FCD binding in the Omicron variant. From Figure 2, it is observed that the P681 H variant produces 12 hydrogen bonds and 22 hydrophobic bonds compared to the N679 K variant’s 12 hydrogen bonds and 12 hydrophobic bonds. In P681 H variant, arginine at the 683 position, which is one of the residues of the cleavage site, forms 4 hydrogen bonds with Asp 153, Asp154, Leu 227, and Gly 229 and 4 hydrophobic interactions with His 194, Asp 228, Arg 185, and Asn 192. These interactions further support the higher binding affinity of the P681 H variant and furin domain.
Although P681 H plays the major role in the binding of furin-FCD of spike protein, it is also clear that the Omicron variant containing both N679 K and P681 H provides the highest binding affinity, which further emphasizes the importance of co-occurrence of both mutations. Even though this co-occurrence increases binding affinity with furin, it negatively affects furin cleavage at the 685-86 position. The negative epistatic impact on the evolution of the spike protein thus must be directed by mutation in both 679 and 681 positions since those mutations alone can provide better fusogenicity than when present together in Omicron. Reports suggested P681 H and N679 K mutations being together results in an optimized FCS, and this optimization might be detrimental to efficient virus transmission due to low fusogenicity. 34
The distance of the furin from the amino acid 685 and 686 would be another important factor that usually defined the enzymatic activity (Supplementary Table 1). Although Omicron shows a close distance to the furin enzyme than wild type, the enzyme activity does not correspond to the results. The catalytic triad of furin is composed of His 194–Asp 153–Ser 368, where serine 368 attacks the carbonyl carbon of Arg685 of the spike protein resulting in cleavage of the peptide bond between Arg 685 and Ser 686 of the spike protein. 40 In our study, we estimated that both Omicron and P681 H variants of SARS-CoV-2 spike protein generated more hydrophobic bonds and less hydrogen bonds with the furin active region (FAR) of the furin enzyme than the wild type and N679 K variants. Delta variant, which has high fusogenicity and linked infection rate, also showed similar number and pattern of interactions just as in Omicron and P681 H variants although the Delta variant has lower hydrophobic interactions than these two variants (Table 4 and Figure 2). Notably, Omicron, Delta, and P681 H variants have a common pattern of amino acid replacement at position 681, with an amino acid of more positively charged side chain, ie, arginine and histidine. 37 The P681R of Delta is however responsible for both spike cleavage and membrane fusion through furin-mediated cleavage showing the importance of amino acid R (Arginine) at that position.41,42
Even though hydrogen bonds provide better protein-protein interaction, the binding between FAR and spike protein was more stable in the variants with more hydrophobic interaction (Table 4). Overall binding patterns by hydrophobic interaction lead to a flexible nature of the binding required after the cleavage activity of furin because the cleaved spike protein needs to initiate fusion with the host envelope for entry. Thus, it would be more optimal for the Omicron variants to have hydrophobicity in that region to achieve better fusogenicity. 43
Besides, even though the wild-type variant has more hydrogen bonds to interact with FAR, it also has proline at the 681 position. In the 681 position, histidine is more beneficial instead of proline, as proline in that position can act as the phosphorylation site for proline-directed kinase. Phosphorylation in that position might be able to decrease the binding and cleavage activities of the furin. 31 Histidine at 681 is also present in Alpha and Mu variants, which is predicted to increase cleavage activity. 34 In addition, Proline in the 681 position promotes glycosylation in this site that is highly associated with the decrease of furin-based cleavage; 44 therefore, the removal of P681 could lead to the loss of the potentially furin-obstructing glycosylation site and associated enhancement of cleavage. 34 Salt bridge interaction is known to generate more binding energy than hydrogen bonds, 45 and in short distances (approximately < 4 A°), it provides additional stability in a protein-protein interaction. 46 It also enhances the nucleophilic attack of proteases. 47 In wild type, variants of spike protein containing asparagine 679 (Asn 679) and proline 681 (Pro 681) has folded in a conformation where no salt bridge non-covalent interaction is present between Arg 685 of the cleavage site and the FAR. P681 H variant has the conformation that produces 3 salt bridge interactions between the Arg 685 (spike) side chain and Asp 191 (furin). N679 K variants also produce two salt bridge interactions between the same residues. But this variant generates a different conformation where amino acid positioning changes compared to the interaction between P681 H and FAR. But not so surprisingly, Omicron variant containing Lys 679 and His 681 has completely different conformation in this location that changes the amino acid position more differently than the previous variants, and it only creates one salt bridge bond between Arg 685 (spike) and Asp 191 (furin) (Figure 3). Even though Delta variants did not create any salt bridge interactions in Arg 685 (spike), we observed that Delta variants created three salt bridge bonds between Arg 681-83 and FAR (Supplementary Figure 2), and the results corroborated with the findings of Cheng et al 42 These three salt bridge interactions enhance the insertion ability of serine 368 of furin catalytic triad and thus promotes cleavage. 42 Omicron variants also has salt bridge bonds in 682 and 683 positions, but the replacement of Arg residue with His at the 681 position resulted in the loss of the remaining salt bridge bond.
Like other serine proteases, a successful catalytic activity of furin catalytic triad requires the stabilization of the intermediate oxyanion hole. This stabilization is performed by the Asn 295 of furin. 48 From Figure 2, we observed that in Delta variants, Arg 681 (spike) has a hydrogen bond with Asn 295 (furin), which enhanced the stabilization of the intermediate oxyanion hole and might result in efficient cleavage formation. Notably, this crucial interaction is not present in other variants.
The Functionality and stability of every protein depend on the folding nature and conformation of the protein and its active domain, 49 which determines its interaction pattern with the target residues. From this study, it is found that the number of bonds and interacting residues in the active pocket got changed although P681 H, Delta, and Omicron variants showed similar number and type of bonds in that region. And near the cleavage site, P681 H and Omicron variants produced a salt bridge interaction with Arg 685 (spike) residue, which may increase the stability of the interaction, and it may also contribute to the increased binding energy. Even though salt bridge bonds provide a better and more efficient mechanism for nucleophilic hydrolysis, they lack the interaction of Arg 681 of Delta variants that leads to efficient cleavage activity of the furin catalytic triad. A study of canonical inhibitors of furin and other serine proteases reveals that those protein inhibitors have the proper conformation and FCS (RRXR) to bind strongly to the furin. Upon binding, they produce an extremely tight and rigid non-covalent interaction, but a lack of efficient cleavage formation leads the furin to be inhibited.50–52
So, the reason behind the low fusiogenicity and less cleavage product formation of spike protein of Omicron variant can be two-fold. First, co-occurrence of P681 H and N679 K mutations in the Omicron variants creates the necessary interactions for the strongest affinity that produces a tight and rigid bond between FCS of spike protein and the FAR of furin. Then, catalytic activity of the furin may become inefficient because of the loss of Arg 681–mediated interaction (one salt bridge bond and hydrogen bond with Asn 295). Therefore, furin catalytic activity gets hindered like product-inhibition type situation and results in the lower fusogenicity of the Omicron.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322231189371 – Supplemental material for Insights into Omicron’s Low Fusogenicity through In Silico Molecular Studies on Spike-Furin Interactions
Supplemental material, sj-docx-1-bbi-10.1177_11779322231189371 for Insights into Omicron’s Low Fusogenicity through In Silico Molecular Studies on Spike-Furin Interactions by Spencer Mark Mondol, Md Hasib, Md. Belayet Hasan Limon and A S M Rubayet Ul Alam in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
We would like to acknowledge the universities for supporting the analysis in the institutions. We would also like to acknowledge and thank Md. Shafi Mahmud for carrying out the molecular dynamics simulation.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MH and SMM: Conceptualization, methodology, validation, formal analysis, investigation, visualization, and writing the original draft.MBHL: Docking analysis with Schrodinger software and writing.MH and ASMRUA: Writing review and editing, supervision, and project administration
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.