
Abstract
COVID-19 caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) had an adverse effect globally because it caused a global pandemic with several million deaths. This virus possesses spike protein that is cleaved or activated by Furin-like protease enzymes occurring by mammalian lung or respiratory cells to enter the mammalian body. The addition of the Furin cleavage site in SARS-CoV-2 makes it a more infectious and emerging virus than its ancestor’s viruses. Phylogenetic relationships of coronavirus spike proteins have analyzed and mapped Furin recognition motif on the tree using bioinformatics tools such as GTEx, KEGG, GO, NCBI, PolyPhen-2, SNAP2, PANTHER, Hidden Markov Models (Fathmm), Phd-single-nucleotide polymorphism (SNP), I-TASSER, Modpred, Phobius, SIFT, iPTREE-STAB, and PROVEAN. During this study, it has been found that in certain regions, Furin SNPs have some relation with the susceptibility to SARS-CoV-2. Whereas in other regions, the effects are very negligible. Finally, our study demonstrates that Furin SNPs have a strong relationship with susceptibility to SARS-CoV-2. As it helps to cleave the spike protein of the virus, thus it can be targeted to inhibit at a particular site to prevent the SARS-CoV-2 from the entrance into the body.
Keywords
Introduction
The current epidemic around the world is caused by a life-threatening virus first detected in Wuhan in December 2019 and rapidly spread throughout the world.1,2 The World Health Organization (WHO) termed this particular virus “severe acute respiratory syndrome coronavirus-2” (SARS-CoV-2) and the associated disease as COVID-19. 1 COVID-19 has been deemed a pandemic by the WHO because of its widespread effects. Around 770 million people had contracted the virus worldwide as of July 2024, according to the WHO, and identified discovery in December 2019, it has been linked to over 6.9 million fatalities. 2 Four types of coronavirus alpha, beta, gamma, and delta are responsible for causing infections in other vertebrae including humans.3,4 Phylogenetic analysis showed that this novel type of SARS-CoV-2 belongs to the coronaviridae family that caused 2 epidemics in the past designated as SARS-CoV and MERS-CoV (the Middle East Respiratory Syndrome Coronavirus), respectively. Since SARS-CoV is a novel virus, even after being included in the coronaviridae family, there are some discrepancies with others. The genome of SARS-CoV-2 has 96.3%, 89%, and 82% similarities with BATCoV, SARS-like CoV, and SARS-CoV, respectively, which confirm the zoonotic origin of SARS-CoV-2. 5
The presence of SARS-CoV-2 can be identified through a range of symptoms in an infected individual, varying from asymptomatic cases to severe illness, and in some cases, leading to fatality. 3 The most common symptoms are fever, cough, and shortness of breath. 3 ,6-8 Some of the other symptoms that have been revealed are weakness, malaise, sore throat, respiratory distress, muscle pain, loss of taste or smell, headache, sputum production, hemoptysis, and diarrhea.3,6 Recent studies have shown that people with diabetes, hypertension, cardiovascular disease, and chronic obstructive pulmonary disease (COPD) have highly severe illness and death.8,9 According to the WHO, the incubation period of SARS-CoV-2 is 2 to 14 days.1,3 Severe acute respiratory syndrome coronavirus-2 is present in respiratory droplets in the air and may spread up to 1 to 2 m and can easily get into someone else through inhalation. 4
A multi-basic Furin cleavage site at the S1/S2 border of the spike protein, which distinguishes this virus from SARS-CoV, was recently discovered by analysis of the SARS-CoV-2 sequence. 10 Furin is a protease that is essential to the SARS-CoV-2 infection cycle because it cleaves viral proteins as the virus particles are assembled. Furin also controls several physiological processes connected to cardio-metabolic characteristics. Owing to different furin gene expressions in tumor tissues, it is crucial in predicting cancer patients’ sensitivity to SARS-CoV-2 and the course of the disease. Candidates for regulating the risk of acquiring these features as well as the vulnerability to severe COVID-19 are DNA polymorphisms in the Furin gene. 11
The furin gene encodes a protein that acts as a Paired Basic Amino Acid Cleaving Enzyme. With the help of transmembrane protease serine 2 (TMPRSS2), Furin facilitates the entry of the SARS-CoV-2 into the host cell after binding through the Furin receptor.12,13 A recent study has identified a Furin cleavage site within the Spike protein of the 2019-nCoV, missing within the other SARS-like CoVs, 14 and it may influence viral morbidity or mortality. Furin cleaves the SARS-CoV-2 spike glycoprotein (S), which is a crucial stage in the viral entry process. S protein is split into S1 and S2 units, with S1 being removed. The remaining viral S2 unit then goes through a conformational reconfiguration that completes the fusion of the viral and cell membranes, allowing the virus to enter the cell, release its material, replicate, and infect other cells. 15 Furin protease may play a crucial role in COVID-19 pathogenicity by increased affinity for its receptor which is present in SARS-CoV-2 spike glycoprotein. 16
Single-nucleotide polymorphisms are present in coding, noncoding, or intergenic regions.17,18 The noncoding and intergenic SNPs may have a silent or light effect, whereas nonsynonymous SNPs (nsSNPs) have a greater effect on protein structure and functions. 16 Since not all reported polymorphisms are harmful, determining how variations affect the protein’s structure, stability, and function is an essential endeavor. 19 Thus, it is essential to understand how nsSNPs negatively affect protein structure and function using various modern molecular biology methods. To date, a huge repository of SNPs reported on NCBI data 20 but going through all nsSNPs and analyzing their impact by lab-based experiments is costly and nonfeasible. However, using computational techniques could be a more effective and efficient alternative for rapid identification and analysis.
Since the Furin protein had a reported cleavage and processing activity of SARS-CoV-2 spike glycoprotein priors to the bind with ACE2 receptor during the cellular entry process 21 and the Furin gene has various nonsynonymous SNP polymorphisms. Thus, there is a possibility that the variants of this gene are involved in the COVID-19 susceptibility among different populations.
Our study aims to investigate the potential association and impact of various Furin SNPs on COVID-19 susceptibility and transmission rates across different geographical locations, employing a range of computational tools. Furthermore, molecular docking and dynamic simulation studies are conducted on significant SNP variants to gain insights into their altered binding affinities and their influence on disease progression.
Materials and Methods
Assessment of Furin’s placement and fluctuation across the genome
For most of the vertebrate species, the Ensembl genome browser (https://asia.ensembl.org/index.html) gathers detailed data on Gene patterns, splice variants, sequence difference, transcriptional control, regulatory function, protein domains, comparative genome research, and disorders. 22 Using this browser, Furin gene protein coding, noncoding region as well as upstream regulatory regions including promoter, enhancer, flanking region analyzed and visualized.
Effects of genetic diversity on the expression of GTEx exons and tissue-specific gene expression patterns
Genotype-Tissue Expression (GTEx) is a website, which may be found at https://gtexportal.org/home/, used to predict inherited disease susceptibility by evaluating tissue-specific gene expression patterns in response to hereditary deviation. This server was used to identify the expression pattern and level of the Furin gene in different tissues.
KEGG, GO, and STRING database analysis of the Furin gene’s molecular pathways
The KEGG database is the web-based tool used for genomic analysis, biological pathways, disease mechanisms and diagnosis, drug discovery, and chemical substances. It may be accessed at https://www.genome.jp/kegg/. The GO (gene ontology) tool is a powerful computational platform used to analyze and investigate the pathways connected with many important genes. It may be accessed at http://geneontology.org/.23,24 Moreover, the STRING (https://string-db.org/) is used for the detection of the protein–protein interaction analysis. In our study, these 2 tools were used to analyze the Furin gene-associated pathways and their functions along with their role in gene ontology for cancer progression.
Characterization and retrieval of SNPs by NCBI and UniProtKB
Sequences of Furin SNP variants were retrieved from dbSNP and the NCBI website (https://www.ncbi.nlm.nih.gov/). The NCBI is a thorough and potent gene browser that represents a vast array of information on genes and protein functions, SNPs, different forms of mutations, and clinical mutational variants. From the UniProtKB database (https://www.uniprot.org/), the amino acid composition of Furin was obtained.
Impact of Furin SNPs on function as determined by SIFT, PolyPhen-2, PROVEAN, SNAP2, Phd-SNP, and PANTHER
Protein folding research in both humans and other species could be conducted with the SIFT (sorting intolerant from tolerant), which is accessible at https://sift.bii.a-star.edu.sg. These tools provide scores ranging from 0 to 1 for alternative splicing of nonsynonymous SNPs25,26 and used to detect the nsSNP variants along with their deleterious effect on protein functions among the sequence of Furin SNP variants we obtained from NCBI.
The Polymorphism Phenotyping Version 2 (PolyPhen-2), accessible at http://genetics.bwh.harvard.edu/pph2/, was used to determine the changeover effect of amino acids on the structural and functional proteins. Hence, the functional effect of SNPs characterizing benign might be damaging, and destructive scores range from 0.0 to 0.15, 0.15 to 0.85, and 0.85 to 1.0, respectively. 27
The PROVEAN is accessible at http://provean.jcvi.org/index.php and was used for investigating the probable effects of amino acid replacements on protein structure and functionality categorizing neutral and deleterious variants. Prediction scores less than −2.5 indicate deleterious variants, whereas scores above it categorized as neutral. 28 Various SNPs in Furin were screened for their effect on protein levels using this scoring function.
According to earlier studies, the investigation of the functional effects of SNPs could be done using Screening for Nonacceptable Polymorphisms (SNAP2), which can be accessed at https://www.rostlab.org/services/snap/. This method assessed the influence of various SNPs on the functionality of proteins. Absolute neutrality and pathogenicity were indicated on the heat map by amino acid substitution scores of 100 (deep blues) and +100 (deep reds), respectively.29,30
The phd-SNP (predictor of human detrimental single nucleotide polymorphism) was a type of support vector machine that predicts the SNPs that affect the structure and function of proteins. It might be viewed at https://snps.biofold.org/phd-snp/phd-snp.html. This service groups the effects of SNPs on illness and assigns a score between 0 and 9. 31 Using protein sequences in FASTA format, substitution changes were analyzed using the PANTHER (protein analysis through evolutionary relationship), which might be accessed at https://www.pantherdb.org. Protein function alterations by SNPs are computed using Substitution Position-Specific Evolutionary Conservation (subPSEC) scores. It was anticipated that a score of 0 is benign and a value of less than −3 is harmful.32,33
Hidden Markov Models (Fathmm) implementation for determining functional SNPs
To compute SNP scores and identify problems connected to the blood, brain, genitourinary, immune, ear, nose, throat, endocrine, eye, heart, and so on, used the powerful application Functional Evaluation of SNPs Utilizing Hidden Markov Models (Fathmm) (https://fathmm.biocompute.org.uk/). 34
Furin SNPs mutational influence on splicing signal
The computational tool Human Splicing Finder (http://www.umd.be/HSF/) can identify splicing motifs in human gene sequences and predict the impact of the disruption of natural splice sites in this study. Human Splicing Finder uses the “position weight matrices” approach with consensus values (CVs) that range from 0 to 100 to identify donor or acceptor splicing sites. Based on the CV score, a score greater than 65 is considered as a donor or acceptor splicing site. Moreover, if the variation score is below 10% and the wild-type sequence scores greater than 65, then the mutation is likely to constitute a novel splicing site. Furthermore, a wild-type sequence score of below 65 and a variant score of more than 10% indicate the likelihood of the occurrence of a unique splice site. 35
Impact of Furin SNPs on secondary and tertiary structures of protein
Protein homology/analogy 2.0 (Phyre2) was a potentially useful tool for analyzing amino acid variants (e.g. nsSNPs), ligand interaction sites, and secondary structures of proteins (alpha-helices, beta-sheets, and coils) used in our study. Another effective and efficient protein secondary structure predictor tool based on SNPs is GOR IV (https://npsa-prabi.ibcp.fr/cgi-bin/npsaautomat.pl?page=/NPSA/npsagor4.html). PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) was a method for anticipating the secondary and tertiary structures of proteins, including coils, alpha helices, and beta sheets.35-37 Our study uses these tools to determine the SNP impact on the structure and function of the protein.
I-TASSER’s analysis of Furin’s 3-dimensional structure
Protein 3D structure could be accurately predicted by using I-TASSER, which is accessible at (https://zhanglab.ccmb.med.umich.edu/ I-TASSER/). Based on the amino acid sequences of macromolecules, I-TASSER computed the spatial coordinates of each atom encompassed into the protein molecules. I-TASSER aims to estimate solvent accessibility, ligand attachment sites, enzyme commission (EC) digits, active sites, and normalized B factor in addition to analyzing iterative protein structure construction.38,39
Molecular docking analysis
The binding affinity test was performed between the significant SNP variants and the spike protein of SARS-CoV-2. For this, the SNP variants sequence was retrieved from the NCBI and 3D structure prediction was performed using the online protein structure prediction server I-TASSER (I-TASSER server for protein structure and function prediction (zhanggroup.org)). The best structure was picked up and docked with the downloaded crystallographic structure Spike glycoprotein of the SARS-CoV-2 from the Protein Data Bank. Molecular docking operation was performed using the ClusPro 2.0 (ClusPro 2.0: protein-protein docking (bu.edu)), HADDOC, PatchDock, AutoDoc, and AutoDoc Vina in a triplicate manner. Ligand and receptor pre-docking preparation were performed by AutoDock 4.2.6 (Download AutoDock4—AutoDock (scripps.edu)) and the result visualizations were performed by BIOVIA Discovery Studio Visualizer (Free Download: BIOVIA Discovery Studio Visualizer—Dassault Systèmes (3ds.com)), PyMOL (PyMOL|pymol.org) and ChimeraX (www.rbvi.ucsf.edu). Graphical representation was performed using the software origin (Download Origin or OriginPro (originlab.com)).
Molecular dynamic simulations
The best-docked protein complexes of rs6226 and rs150925934 are further performed molecular dynamic simulations with the GROMACS compared with the standard Dock of Furin. Using a GROMACS-2019 software package (GNU and CHARMM37 force field) with Furin as the receptor and SARS-CoV-2 spike glycoprotein as the ligand complex as starting coordinates for a 100 ns all-atom molecular dynamics simulation. The transferable intermolecular potential was used to solvate each ligand-protein combination inside a cubic box of 100 × 100 × 100 Å. The 3-point (TIP3P) water model was used as it allowed for a minimum of 10 Å of marginal distance between each side of the 3D box and the protein. With the aid of the CHARMM General Force Field (CGenFF) tool, the CHARMM force field parameters for the ligands under investigation were automatically created. The protein residues were allocated to their standard ionization states at physiological settings (pH 7.0) under periodic boundary conditions implementation. Then, the entire complex was neutralized by adding enough K+ and Cl− ions using the Monte-Carlo ion-placing method. The 1000 kJ/mol nm2 force constant was used to constrain all heavy atoms and maintain the original protein folding during the 3 stages of the MD simulation. After, a successful energy minimization and equilibrium of the system the MD simulation is run for the 100 nanoseconds. GROMACS build tools were used to postanalyze and compute the MD trajectories comparative data such as RMSD, RMSF, radius of gyration (Rg), and binding energy calculation.
Evaluation of SNPs’ effects on miRNA function and the emergence of severe disease using PolymiRTS
The impact of polymorphism on miRNA seed areas and target profiling was estimated using PolymiRTS (http://compbio.uthsc.edu/miRSNP/). 39
Results
Furin gene sequence and assumption of protein
The gene Furin is positioned on Xq26.1 and has 16 exons (Figure 1). Moreover, Furin has many reported structural changes including insertions, deletions, copy number variations (CNV), duplications, and tandem duplications demonstrated in Figure 2. Gtex also contributed to the determination of the normalized effect size (NES) of several tissues, for example, whole blood (WHLBLD), adipose-subcutaneous (ADPSBQ), artery-tibial (ARTTBL), lung (LUNG), nerve-tibial (NERVET), skin not sun-exposed (SKINNS), skin sun-exposed (SKINS), thyroid (THYROID), and skin not sun-exposed (SKINNS). However, Furin is highly expressed in the liver, whole blood, kidney, lung, thyroid, and spleen; and weakly expressed in the small intestine, cervix, brain, and adrenal gland (Figure 3B).

Furin gene location from the Ensembl genome browser. It is located on Chromosome 15, q21.1 position.

Furin gene detailed mapping in the human genome along with the presence of exon-intron location, their size, and type of SNP presence. Moreover, the probable Pfam domain, SMART domains, prints domain, and Superfamily domains are also shown here.

Furin’s positioning in Gtex was accompanied by the eQTL mapping. (A) Furin’s expression profiles are affected by genetic polymorphisms that are classified by expression quantitative trait loci (eQTL); (B) Comparing the expression levels of furin in various tissues. A variety of tissues are depicted, each with varying degrees of furin expression. The liver and lung exhibit elevated levels of furin expression, while the brain and spleen show lesser amounts; (C) Expression of the furin exon across multiple tissues with a median read count for each base score. To determine the expression of each gene, read count measures the number of reads that map, or align, to the gene (using RNA-seq). A transcript’s length (longer transcripts possess greater read counts at the same expression level) and the total number of readings are 2 factors that can affect raw read counts.
SNPs screening for Furin
NCBI dbSNP database retrieval found a lot of variants such as intronic (4559), missense (635), synonymous (413), in-frame deletion (5), insertion (2), and noncoding transcript (1825), and exons (16) (Figures 4A and 5). However, we limited our analysis to those SNPs having an MAF > 0.001, resulting in 182 SNPs retrieval from the 1000 genome project (Supplemental Table S1) which includes 114 intronic, 8 synonymous, and 6 missense SNPs. Among these, only 29 SNPs had significantly distinct frequencies among the populations of different geographical locations (Table 1). Out of these 29 SNPs, one intronic SNP, rs11073956, demonstrated a noteworthy distinction between Southeast Asians and other populations. In addition, there were differences between Africans and other populations in 10 intronic (rs6496734, rs8024061, rs8030001, rs28509319, rs28580438, rs57509730, rs59312952), two 3 prime UTRs (rs6228, rs6229), two 5 prime UTRs (rs8027907, rs8033171), 2 regulatory regions (rs58147987, rs59856426), and 2 intergenic SNP (rs74037504, rs74037505). Moreover, three 3 prime UTR SNPs (rs1436643, rs78192020, rs79596457) and 7 intronic SNPs (rs4526996, rs28402904, rs75000622, rs78569252, rs113579461, rs111417372, rs4526996) showed a distinct frequency in the European population compared to the other populations (Figure 6).

(A) Furin SNPs in NCBI database. (B) KEGG and GO pathway analysis results. FURIN protein handles the activation of these proteins. Beta-secretase 1, beta-site APP cleaving enzyme 1 (BACE1), is an aspartic acid protease important in the formation of myelin sheaths in peripheral nerve cells. A disintegrin and metalloprotease 17 (ADAM17), also called TACE (tumor necrosis factor-α-converting enzyme) is understood to be involved in the processing of tumor necrosis factor-alpha (TNF-α) at the surface of the cell. Brain Protein I3 (BRI3) is a protein which takes part in tumor necrosis factor-alpha (TNF)-induced cell death. A disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) can cleave all the large chondroitin sulfate hyaluronan-binding proteoglycans (CSPGs). B-type natriuretic peptide (BNP) is one of several proteins that help regulate blood circulation throughout the body. gp160 is a human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein responsible for the integration of the virus into the host cell. (C) Significant SNPs distribution in different continents.

Docking visualization of the reference FURIN receptor and Interacting Ligand part of Spike glycoprotein of SARS-CoV-2. A. Detail view of FURIN-Spike protein docking. Here, the gray color of FURIN is coil, cyan and cornflower blue color represent the helix and strand, respectively. B. Zoom view of interacting area with associated amino acid take part in interactions. C. Detail view of H bond generating surface of interacting domain of receptors. D. Hydrophobic and hydrophilic area visualizations of the FURIN receptor. E. 2D diagram of the receptor-ligand interacting pattern with amino acid residues and bond generation visualizations.

RS626 and RS150925934 FURIN SNP Docking with SARS-CoV-2 Spike Glycoprotein. A(I) is the broad view of the spike glycoprotein docked with the RS626 SNP of FURIN. A(II) Zoom view of the interacting area. A(III) Detailed view of the docked area with associated amino acid next to the binding region. B(I) Broad view of the s protein dock with FURIN SNP RS150925934. B(II) Insight view of the docking area, B(III) Detail view of the interaction area with the adjacent amino acid of FURIN RS150925934 SNP.
The 1000 Genome Project indicates that various populations have varied frequencies of Furin SNP variations. AMR stands for American, EAS for East Asian, SAS for South Asian, AFR for African, and EUR for European.

Functional effects of SNP prediction on stability and function of proteins
The in-silico tools predicted that rs6226 andrs150925934 are deleterious and destabilize Furin and rs16944971, rs116359616, and rs148110342 were predicted as tolerated (Table 2). To investigate the effects of rs6225, rs4405529, rs4526996, rs28580438, rs57509730, rs73489557, rs73489568, rs143669943, rs376483565, and rs557382838 on the splicing process, Furin function has demonstrated that rs28580438 produced new acceptor sites, new exonic splicing enhancer (ESE) sites, and exonic splicing silencer (ESS) sites broken. Thus, rs4405529 and rs37648356 cause an ESS site to break and an ESE site to break, respectively. While rs6225 results in a broken ESE site, rs143669943 leads to a new donor site, and rs557382838 results in a new donor site, an ESE site, and an ESS site. Moreover, rs57509730, rs73489557, and rs73489568, only created a new acceptor site, and rs4526996 resulted in ESE site break-down and Fresh ESS site creation in turn (Table 3).
Prediction of deleterious nsSNPs from various tools.

HSF splice site results.

Secondary structure of Furin SNPs analysis
PSIPRED and GOR IV showed that Furin protein forms the 3 types of structure where alpha helix 17.13%, extended strand 22.92%, and random coil 59.95% (Figure 7B and C). Uniprot also showed strong functional impacts of Furin SNPs (Figure 7A).

(A) Uniprot’s functional analysis of Furin SNPs. A small subset of Furin SNPs was demonstrated to be unclear, while most were shown to represent anticipated sequences. (B) GOR IV predicted the FURIN Protein structural information. (C) PSIPRED results show secondary structure information of FURIN protein.
Protein tertiary structure prediction and investigation based on Furin SNPs
From the I-TASSER server, we have found that G617C and R464W have a different structure than the wild type. G617C has different ligands that are ZN and RAT. Again, R464W also has different ligands that are MG and TRP (Table 4).
I-TASSER result of wild, G617C, and R464W.

Molecular docking analysis of Furin SNPs rs6226 and rs150925934 with SARS-CoV-2 spike protein
Our analysis found rs6226 and rs150925934 as the most deleterious, nonsynonymous significant SNP variants of the Furin gene among all other SNPs of it. Thus, Molecular Docking operations and visualizations are performed where SNP Furin binding affinity with spike glycoprotein is compared with the reference Furin structure and analyzed. Single-nucleotide polymorphism variants show significant structural variations and interaction patterns than the wild-type Furin (Figures 5). Besides, binding affinity and energy levels are also found at elevated levels (Figure 8). Single-nucleotide polymorphism variants can be more tightly bound with the spike protein and may have an impact on the pathogenesis COVID-19.

FURIN SNP shows an elevated level of binding affinity toward spike glycoprotein of SARS-CoV-2 shows an effect in interaction patterns and COVID-19 progressions.
Molecular dynamic simulations
In only RMSD analysis of Furin and its associated SNP Furins, the normal Furin exhibits less flexibility compared to the rs150925934 and rs6226. After ligand fit protein model RMSD analysis, the rs6226 model represents its rigidity while binding with the ligand. However, the rs150925934 model is bound to its ligand, and a huge number of flexibility movements were recorded throughout the 100ns simulations compared to the standard Furin. The RMSF of the 3 modes represent that the Normal Furin residues are more rigid and less prone to deviate. But, the rs150925934 model residues show the most deviations compared to the standard. The rs6226 model deviations are comparatively lower than the rs150925934 (Figure 10). According to the radius of gyration, there is almost a plateau graph for the protein-ligand complex. But the normal furin has the more compact structure, whereas the rs6226 is the least compact structure during the time frame periods of simulations, and the rs150925934 represents moderate structural configurations.

FURIN protein-protein interaction analysis. Here, A. Represents gene co-occurrence analysis; B. Represents networks of FURIN with other interacting nodes; C. Represents gene co-expression analysis in humans and other organisms.

Molecular dynamic simulation of normal Furin, compared to the rs6226 and rs150925934. Here, A, B, C, and D are the RMSD values of the 3 protein-ligand complexes. E, F, and G are the RMSF values of the 3 protein-ligand complexes from different perspectives. In addition, in figures C, D, E, and F black color is labeled for standard FURIN, Red color represents rs150925934, and Green color represents rs6226. Moreover, in Figure G Red color represents rs150925934, Green represents rs6226 and Blue represents the normal FURIN.
There is a great deviation in the binding energy calculations of the interaction molecules to the ligand binding area. In standard Furin, the pocket atoms show elevated binding forces. Whereas, in rs6226 more amino acid molecules are involved in bond configurations with elevated and moderate energy in some cases. But, in rs150925934 with the significantly elevated binding energy of the molecules, there was some significantly lower binding energy molecules involvement in bond interactions along with the more molecular amino acid involvement in the docking area (Figure 11).

Postsimulation analysis of rs6226 FURIN, rs150925934 FURIN and normal FURIN, Radius of Gyration in A and B, Binding energy calculations in C, D, E which are rs6226, rs150925934 and standard FURIN, respectively.
According to the Binding energy analysis, both the SNP variants of Furin (rs6226 and rs150925934) exhibit more stability compared to the wild-type variants. However, the rs150925934 is the more stable with its elevated binding energy, and significantly negative electrostatic and solvation energies. The total Energies of the wild-type Furin is 10 652.06 Kcal/mol whereas the SNP Furin of rs6226 and rs150925934 is −20017.98 Kcal/mol and −23010.78 Kcal/mol which is around 2 times greater (Table 5).
Binding free energy calculation of Normal Furin complex compared to the SNP Furin rs6226 and rs150925934 after MD simulations. Here, DIHED, VDWAALS, ESURF, GSOLV, EEL, EGB, and GGAS are the dihedral energy, van der Waals interactions, surface energy, solvation-free energy, electrostatic energy respectively, generalized born solvation energy and gas phase energy, respectively.

Evaluating the miRNA profiles regarding Furin SNPs across various populations
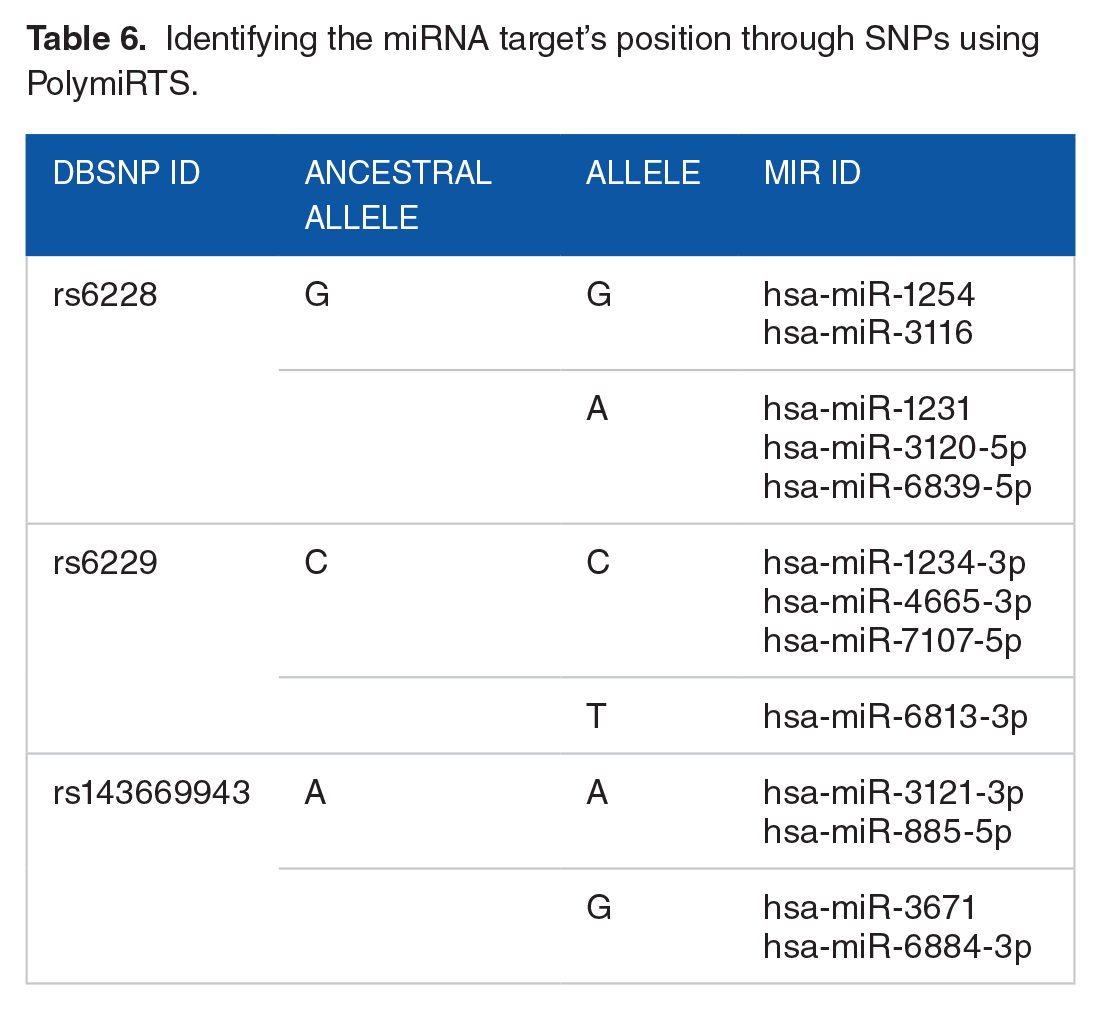
By affecting miRNAs binding to Furin transcripts, Furin SNPs might have an impact on the functionality and biogenesis of miRNAs. Three SNPs that affect miRNA target sites—rs6228, rs6229, and rs143669943—have been identified by PolymiRTS as either disrupting or establishing new target sites (Table 6).
Identifying the miRNA target’s position through SNPs using PolymiRTS.
Discussion
Deciphering population-specific responses to SARS-CoV-2 is crucial for addressing its morbidity and mortality. Severe acute respiratory syndrome coronavirus-2 can infiltrate organs via proteins like Furin, a protease more widely distributed than TMPRSS2. A Furin cleavage site enhances viral spread, while its deletion reduces replication and disease severity.40,41 In kidney cells, Furin inhibitors lower viral generation and cytotoxicity. 42 Variability in Furin SNPs may influence susceptibility, with GTEx showing high Furin expression in adipose, tibial artery, lung, tibial nerve, unexposed skin, thyroid, and whole blood (Figure 3A to C).
Various research showed that patients with comorbidities such as the cardio metabolic continuum years before the onset of diabetes, hyperinsulinemia, hypertension, hyperlipidemia, and obesity,43,44 chronic kidney disease, smoking, diabetes mellitus, and COPD may be more susceptible to SARS-CoV-2 due to elevated Furin expression. However, any alteration in Furin expression levels due to SNPs, splicing, PTM, and transcription processing may make people more susceptible to transmitting COVID-19. The KEGG pathways, protein-protein interaction analysis and gene ontology analysis reveal that Furin is involved in various crucial pathways. Various nervous system proteins like ADAM1, BACE1, and BRI3 are activated by the Furin protein (Figures 4B and Figure 9).
NCBI dbSNP database retrieved data revealed that only 29 SNPs out of 7364 (Figure 4A) in Furin, including 20 intronic, five 3 prime UTR, two 5 prime UTR, and 2 regulatory regions had an MAF >0.001 with varying rates across diverse groups. From them, several nsSNP analyzing tools predicted rs6226 and rs150925934 as deleterious. Remarkably, the findings of the HSF analysis of the SNPs have shown that the splicing process is impacted by rs6225, rs4405529, rs4526996, rs28580438, rs57509730, rs73489557, rs73489568, rs143669943, rs376483565, and rs557382838 (Table 3). The data obtained from HSF suggested that the SNPs that disrupted splicing processes might potentially impact the production of Furin of over 3 prime UTR SNPs, which could then impact the cell entry of SARS-CoV-2. Interestingly, the MAF of those splicing-influencing SNPs was noticeably greater than that of 3 prime UTR SNPs.
The molecular docking analysis of Furin SNP especially rs626 and rs150925934 which have deleterious effects found before shows a significant increase in the binding affinity with the SARS-CoV-2 spike glycoprotein compared with the wild-type furin. The amino acid interacting regions also show significant variations. Standard Furin protein’s docking region contains the amino acid GLN (A:129), ARG (A:130), HIS (A:405), LYS (A:419), PRO (A:403), ALA (A:404), GLY (A:417), LYS (A:402), THR (A:400), ARG (A:581), SER (A:401), GLN (A:399), ARG (A:418), VAL (A:398), LEU (A:578), and many more which form the interaction type of van der Waals, salt bridge, attractive charge, conventional hydrogen bond and pi-sulfur bond (Figure 6). Furin SNP rs626 interaction regions with S protein have amino acid of GLN391, ARG387, PHE261, LEU252, TRP360, LEU227, GLY 103, TYR393, HIS 253, PRO264, GLU96, ALA173, LEU46, HIS272, SER41, and VAL60. Whereas another deleterious SNP rs150925934 interaction domain with s protein hold the amino acids of PRO575, TRP 576, SER574, LEU573, CYS283, GLY287, PRO213, SER209, GLN206, VAL207, ARG198, ALU179, ALA178, GLY180, ASP195, GLB 277, ALA278, and SER183 (Figure 7). Besides, the binding energy level is elevated in SNP variants of rs626 and rs150925934 with the values of −1235.6 and −1456.9 while the wild-type furin binding energy found is −618.8 (Figure 8). Thus, a major impact in structural changes because of SNP also affects its interaction pattern and progression of SARS-CoV-2.
Out of 29 SNPs, one intronic SNP was found to differ significantly between Southeast Asians and other populations. In addition, there were differences between Africans and other populations in 10 intronic, two 3 prime UTR, two 5 prime UTR, 2 regulatory regions, and 2 intergenic SNP. Furthermore, compared to other populations, the European population displayed a distinct frequency for 7 intronic SNPs, including 3 prime UTR SNPs (Figure 4C). This implies that the susceptibility and furin SNPs are related.
Further secondary structure analysis using GOR IV revealed that the Furin protein forms 3 distinct types of structures: random coils (59.95%), extended strands (22.92%), and alpha helices (17.13%). G617C and R464W were predicted by PSIPRED to be the probable domain and beta-strand site, respectively. Consequently, variations in Furin structure may affect COVID-19 cell entrance. PolymiRTS was used to examine how the profiles of miRNAs changed in response to Furin SNPs in various groups. Three SNPs, rs6228, rs6229, and 143669943, were predicted by PolymiRTS to have an impact on the miRNA profile in several populations with an MAF > 0.001.
Conclusion
This study reveals that Furin SNPs, particularly rs6226 and rs150925934, increase susceptibility to SARS-CoV-2 by enhancing the binding affinity between Furin and the virus’s spike protein, which may facilitate viral entry and replication. The distribution of these SNPs across populations suggests genetic factors may contribute to varying COVID-19 susceptibility. Our findings support the potential for Furin-targeted therapies tailored by population-specific genetic profiles. Further studies are essential to validate these insights and explore precision treatments for COVID-19.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322241306388 – Supplemental material for Variations in Furin SNPs, a Major Concern of SARS-CoV-2 Susceptibility Among Different Populations: An In-Silico Approach
Supplemental material, sj-docx-1-bbi-10.1177_11779322241306388 for Variations in Furin SNPs, a Major Concern of SARS-CoV-2 Susceptibility Among Different Populations: An In-Silico Approach by Md Nasir Uddin, Md Arzo Mia, Yasmin Akter, Mohammad Al-baruni Chowdhury, Md Hadisur Rahman, Hafsa Siddiqua, Umme Salma Shathi, Abdullah Al-Mamun, Farida Siddika and Lolo Wal Marzan in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was partially supported by the Research & Publication Cell of the University of Chittagong, Bangladesh (Ref. 342/2021; the fiscal year 2021-2022) which was done in the “Microbial Genomics and Metabolic Engineering Laboratory” of the Department of Genetic Engineering and Biotechnology, University of Chittagong. We dedicate this research to the courageous participants and martyrs of the “Anti-Discrimination Movement” in Bangladesh during July to August 2024. Their strong commitment and sacrifice in the pursuit of fairness and justice inspire us all and pave the way for building a brighter future for our nation.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
This work is the culmination of collective intellectual efforts from the entire team, with each member contributing varying degrees of expertise to the analytical methods, research concept, experimental design, and manuscript preparation.
Consent for Publication
All authors have read and approved the final version of this manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.