
Abstract
The increased transmissibility of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has generated variants of concern (VOCs) throughout the pandemic, responsible for waves of cases worldwide. To monitor mutations in the S gene of SARS-CoV-2 in different variants, we evaluated 1497 individuals with COVID-19 in western Amazonia in the period April 2021 to July 2022. The epidemiological and clinical data of the individuals were collected; subsequently, the samples were extracted using a commercial kit, the viral load was assessed, and viral genomes were sequenced. We analyzed the quality and mutations of the genomes and maximum likelihood phylogenetic inference. However, 3 main clusters were observed, referring to Gamma (52.91%), Delta (24.38%), and Omicron (20.38%) VOCs with wide distribution in all health regions of the Rondônia state. Regarding the vaccination profile, there was a higher percentage of unvaccinated and partially vaccinated individuals, with more representatives by the Gamma variant. A total of 1412 sequences were suitable for mutation analysis in the S gene region. The Omicron VOC showed 38 mutations, with the Delta and Gamma variants with 16 and 17, respectively. The VOC Omicron and Gamma shared 4 mutations E484K, H655Y, N501Y, and N679K with high frequency, and Delta and Omicron 2 mutations (T478K and T95I). Regarding the comparison between the frequency of mutations for each variant concerning the vaccination groups, there were no changes in mutations for each group. In conclusion, the study showed a temporal increase in mutations and subvariants for characterized strains. Furthermore, the vaccination profile did not impact significant changes in the mutational profile yet remains a determining factor for severe disease.
Introduction
The emergence of mutations in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome is a determining factor in the emergence of epidemic waves. It is important to point out that even though it has a genetic-revision mechanism, 1 the virus has not been able to contain more than 25 000 mutations already reported. 2 Mutations may enhance transmissibility, pathogenicity, and host immune escape; however, some substitutions may not change the viral phenotype. 3
Among the regions of the viral genome, the gene encoding the Spike (S) protein continues to be a major center of modification. The Gamma and Delta variants, previously circulating variants of concern (VOCs), and the more recent Omicron variant have as their main characteristic the gradual increase in mutations in the Spike region and the subsequent increase in the number of cases of the disease. 4
The VOC Gamma (P.1), identified in November 2020 in Amazonas State/Brazil,5,6 was responsible for the second wave of infections and deaths, and brought as characteristic mutations: (a) L18F, T20N, P26S, D138Y, and R190S in the N-terminal domain (NTD); (b) K417T, E484K, and N501Y in the receptor-binding domain (RBD); (c) D614G and H655Y in the C-terminus of S1; and (d) T1027I and V1176F in S2. The K417T, E484K, and N501Y mutations were of greater concern due to their location on the angiotensin-converting enzyme 2 (ACE2) contact surface, which may provide escape from neutralizing antibodies. 7
The Delta (B.1.617.2) variant was reported in India in October 2020, possessing 11 characteristic mutations being T19R, T95I, G142D, Δ156-157, R158G, L452R, T478K, D614G, P681R, and D950N in the Spike protein, showing high case numbers; however, no significant impacts in severe cases.8-10
The Omicron variant (BA) was first described in Africa in November 2021 and has the largest number of mutations described among all the variants, accumulating approximately 34 in the S protein alone. However, 2 of these mutations (N501Y and Q498R) can increase the transmissibility of the SARS-CoV-2 with the capacity to intensify the interaction with the ACE2 receptor. 11
Considering the economic, social, and health impacts caused by virus mutations, this study aims to show the mutations in the gene encoding the Spike (S) protein of SARS-CoV-2 in different variants, comparing the result between vaccinated and unvaccinated populations in western Amazonia.
Materials and Methods
Ethical aspects and biological samples of SARS-CoV-2
This study was conducted in the Laboratório de Virologia Molecular at Fiocruz/RO, with approval from the Research Ethics Committee of the Research Center for Tropical Medicine of Rondônia-CEPEM/RO 4.000.086 and was performed in accordance with the ethical principles stipulated by the 1975 World Medical Assembly and the Ministry of Health (Resolution 466). All experiments were performed in accordance with the relevant guidelines and regulations, and were exempt from informed consent requirements due to their retrospective design.
A total of 1497 SARS-CoV-2 positive individuals were selected by convenience from primary care clinics and reference centers in different municipalities of the Rondônia state from April 2021 to July 2022 for genome sequencing. SARS-CoV-2 diagnosis was conducted in Laboratório Central de Saúde Pública de Rondônia (LACEN/RO) by real-time quantitative reverse transcription PCR (RT-qPCR) with One-Step/COVID-19 kits (IBMP, Brazil). Epidemiological data and vaccination status were collected from medical records in the GAL/RO, SIVEP-Gripe, and E-SUS databases.
Extraction of Viral RNA
A total of 140 µL of samples were collected with combined swabs; then, viral RNA was extracted using QIAamp® Viral RNA Mini Kits (QIAGEN, Germany) according to the manufacturer’s instructions. The RNA was eluted in 60 µL of Elution Buffer (AVE) for viral load and inference tests.
SARS-CoV-2 amplification and sequencing
A Multiplex One-Step RT-qPCR assay for the detection of SARS-CoV-2 as developed by Queiroz et al 12 was used for the viral load measurement. The sequencing of SARS-CoV-2 complete genome was performed with the support of the FIOCRUZ Genomic Surveillance Network at Fiocruz unit in Manaus, Amazonas State. Samples with Ct values less than 25 were selected for high coverage. Nucleotide sequencing was performed using Illumina MiSeq or NextSeq 1000 platforms using COVIDSEQ Kit (Illumina, San Diego, CA, USA), as previously reported. 13
Mutation analysis
SARS-CoV-2 genomes were classified into lineages using the available software Pangolin (version v4.2, pangolin-data version v1.18.1.1), 14 and mutations were analyzed with Nextclade Beta (version 2.13.0). 15 Nextclade implements several quality controls (QCs) to flag potentially problematic sequences due to errors during sequencing or assembly. Sequences that produced mediocre and poor metrics were excluded from mutation analyses.
Maximum likelihood phylogenetic reconstruction
High-quality (>29 kb) whole reference genomes (<1% of N) of VOCs Gamma, Delta, and Omicron sampled in Brazil (n = 47) were downloaded from the GISAID EpiCoV database on September 11, 2022. 16 The sequences generated in this study (n = 1412) and the retrieved sequences were aligned using MAFFT v.7.487. 17 The best model of nucleotide substitution was measured (GTR + G + I) using ModelFinder, 18 and the phylogenetic tree was reconstructed using the maximum likelihood method in the program IQ-TREE v.2.1.3. 19 Branch support values were obtained using Ultrafast Bootstrap with 1000 replicates. The tree was visualized and edited with FigTree v.1.4.4. 20
Statistical analysis
Descriptive analyses were represented through central tendency and dispersion measurements. A chi-square test was used for statistical inference with a significance level of 5% (P < .05). Statistical analysis was performed, and graphics were generated using R software v4.0.3.
Results
A total of 1497 samples from individuals infected with SARS-CoV-2 from April 2021 to July 2022 were analyzed. All samples were characterized as to variant and subvariant; Figure 1 demonstrates the maximum likelihood phylogeny inference in relation to the classification of the major clades of the identified Gamma (P.), Delta (AY.), and Omicron (BA.) variants and their respective subvariants.

Maximum likelihood phylogenetic tree showing 1412 sequences with “good” quality in NEXTCLADE obtained in this study and 47 reference genomes retrieved from GISAID.
The Gamma variant showed the highest numbers corresponding to 52.91% (792/1497), followed by the Delta variant with 24.38% (384/1497) and the Omicron VOC with 20.38% (321/1497) of the cases reported in this cohort. The map demonstrates the geographical distribution of the variants in the 7 health regions of the state of Rondônia with a marked prevalence of Gamma in the Madeira-Mamoré Health Region, Delta in the Jamari Valley (81%) and Guaporé Valley (72%) regions, and Omicron with greater distribution in the Zona da Mata (43%) (Figure 2).

Dispersion map of cases per variant in the 7 health regions in the state of Rondônia.
The Omicron VOC had the highest number of subvariants (n = 19), followed by Delta (n = 12) and Gamma (n = 5) circulating in the state of Rondônia during the study period.
In the first genomic surveillance periods, in mid-April, there was a prevalence of VOC Gamma, where the subvariants with the highest number of cases were P.1 and P.1.4, with 84.60% (670/792) and 11.87% (94/792). In August 2021, with the emergence and circulation of VOC Delta in the state, the subvariants with the highest number of infected individuals were AY.99.2 with 46.61% (179/384) and AY.43 with 46.09% (177/384) of cases. VOC Omicron was first detected on December 20, 2021 and has since been the only VOC detected through July 2022, with BA.1 showing 47.04% (151/321) and BA.1.1 with 15.89% (51/321) of characterized cases (Figure 3).

Area plot showing the distribution of subvariants during the study period.
The viral load of the individuals presented a median of 7.33 (SD = 1.07) log10 copies/mL. Regarding the period of viral load detection, the study population showed a median of 4 days of symptoms (SD 2.7, Min 1 day and Max 16 days). There was no significant difference when each detected variant was individually evaluated.
Regarding the vaccination profile evaluated, 61.92% (927/1497) belong to the unvaccinated and partially vaccinated group (includes unvaccinated and first dose only) and 38.08% (570/1497) to the fully vaccinated group (includes second dose and boosters). Within the unvaccinated and partially vaccinated group, VOC Gamma had the highest number of infected individuals with 74.76% (693/927). Within the fully vaccinated group, the prevalence of the VOC Omicron was 44.74% (255/570) in relation to the period studied.
Among the infected, the priority groups (age group > 60 years and with comorbidities) had a higher vaccination coverage. Among the groups listed in Table 1, the analyses showed that vaccination was a significant protective factor against hospitalization.
Profile of vaccination groups with respect to severity, variants, and epidemiological data.

Abbreviations: CI, confidence interval; VOC, variants of concern; ORc, Odds Ratio Crude; ORad, Odds Ratio Adjusted.
and bold indicates significance value.
A total of 1412 were quality for mutation analysis according to Nextclade metrics, and mutations with a frequency above 2% in the S gene region were recorded and analyzed. The Delta variant had 16 mutations in this region, Gamma had 17, and the Omicron variant had 38 mutations in this gene. It was possible to verify that the VOC Omicron and Gamma shared 4 mutations with high frequency, being E484K, H655Y, N501Y, and N679K, and to a lesser extent the Delta and Omicron variant shared the T478K and T95I mutations (Figure 4).

Mutations located in the S gene region in the Gamma, Delta, and Omicron variants.
Figure 5 shows the mutation profile for each variant in relation to the unvaccinated/partially (no dose or first dose only) and fully vaccinated (second dose or booster doses) groups. The variants were circulated at different times from the inclusion of vaccination doses and in the evaluation, it was possible to see that the vaccination profile had no impact on the change in mutational profile.
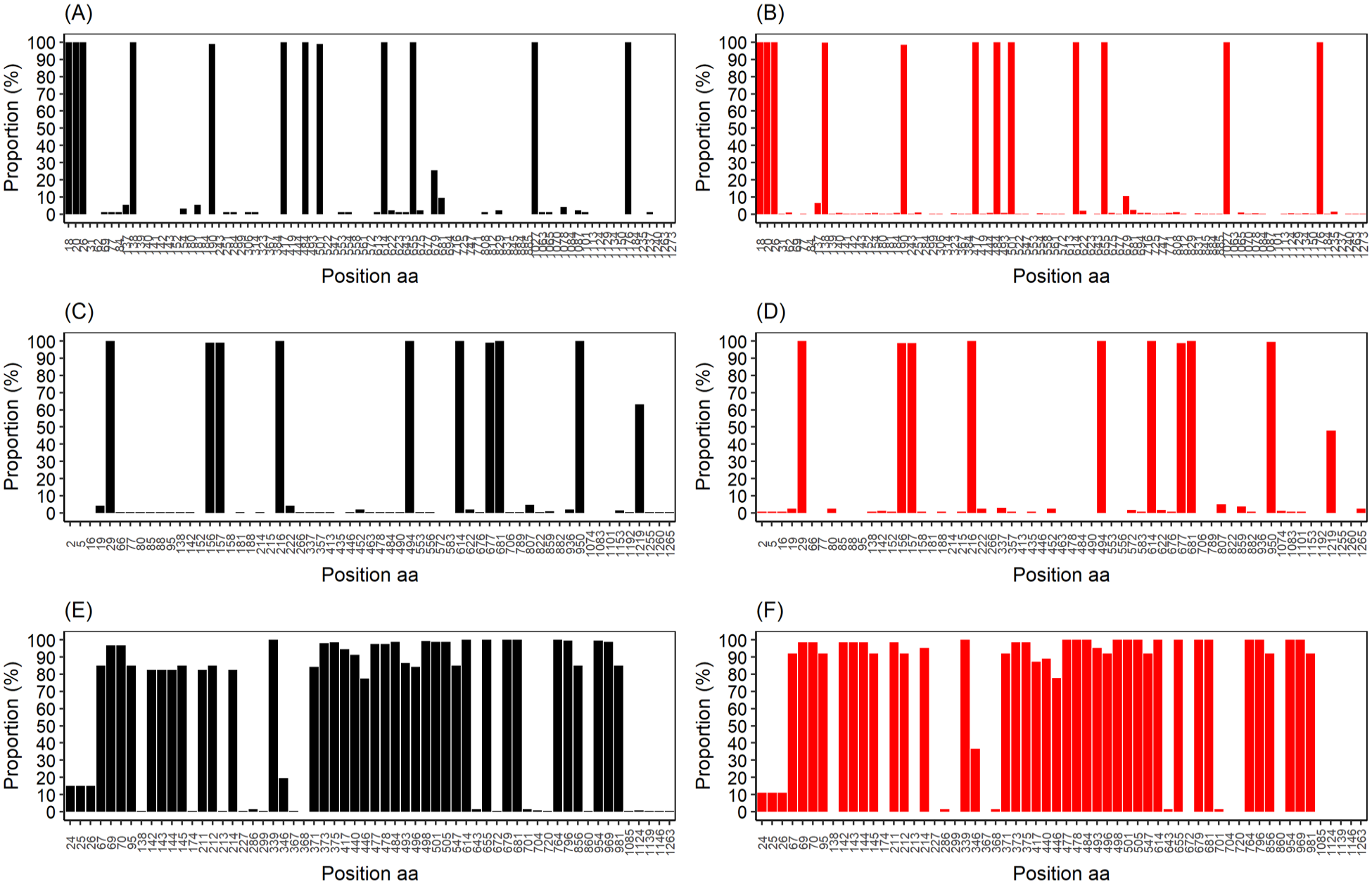
Immunization groups compared with proportions of mutations for each variant. (A) Gamma vaccinated. (B) Gamma nonvaccinated/partially. (C) Delta vaccinated. (D) Delta nonvaccinated/partially. (E) Omicron vaccinated. (F) Omicron nonvaccinated/partially.
Discussion
The distribution of the variants detected shows the temporal context of SARS-CoV-2 infections, demonstrating 3 distinct periods of infections in the state of Rondônia, initially characterized by the dominance of VOC Gamma, followed by Delta and finally Omicron.
The phylogenetic tree of the study corroborated with the clade structure currently proposed for SARS-CoV-2, 15 showing a large accumulation of Gamma variant cases.21,22 The Gamma variant was quickly replaced in Rondônia by the Delta variant; however, it was not responsible for new waves of cases like those previously reported.23,24 A smaller number of individuals appear related to the Omicron variant reported lately as an alert. 25
In Rondônia, the rate of sequenced samples was satisfactory in all health regions compared with other regions of the country, keeping genomic surveillance levels active.26,27 The replacement of variants possibly demonstrates the contribution of the vaccine spectrum in the state, considering that the Madeira-Mamoré region is responsible for housing the largest number of inhabitants and only reflected the highest prevalence for Gamma compared with Delta and Omicron during all analyzed periods. According to the sample selection criterion, which is based on the quality of the genomic sequencing, the viral load level of the study population was expected to be high, as it proved to be, because it is the main influencer on the success of the sequencing.28,29
We observed that fully vaccinated individuals composed most of the individuals who became infected with Omicron, as the circulation occurred after the period when vaccination was already fully included in the study population. 30
Several studies revealed that VOC Omicron has a higher transmissibility compared with the previous variants and, thus, demonstrates the potential of the variant in the vaccine escape.31-34 However, the results showed that individuals with full vaccination had a lower risk of hospitalization compared with those who were not vaccinated or who received only 1 dose. In fact, vaccines against COVID-19 have direct efficacy in decreasing the transmissibility and especially the severity of the disease.35-37 In addition, other studies indicate the booster dose is essential in controlling the disease.38-40
Some mutations already described may promote resistance to immunization, 41 but according to our evaluations, there were no significant changes in the frequency of mutations in the S gene compared with the characterized groups (vaccinated and unvaccinated/partially).
The D614G mutation reported since the beginning of the pandemic 3 showed high prevalence in the Gamma, Delta, and Omicron VOCs in the analyses performed. This mutation is closely linked to a higher affinity in the binding of variants for the ACE2 receptor, due to the presence of the alteration in the RBD conformation, thus increasing its transmission rate.42,43 In addition, other signature mutations, such as E484K, H655Y, N501Y, and N679K, were shared between the Omicron and Gamma VOCs, linked to neutralizing antibody resistance to SARS-CoV-2, suggesting that viral evolution is also linked to vaccine-resistant mutations. 44
In conclusion, the data demonstrated the temporal dissemination of the 3 main VOCs characterized in the state of Rondônia, in the Western Amazon region in the year 2021 (April) to 2022 (July), verifying the accumulation of mutations for each variant, resulting in an increase in the number of subvariants. Moreover, the immunization profile did not impact the change in mutational profile but was a determining factor for the control in the evolution to severe cases.
It is important to note that other mutations with lower frequency were described in this study in the different periods. These mutations presented with lower frequency may become possible signature mutations with potential for higher virulence or transmissibility rate as occurred with VOC Omicron, especially with mutations in the S gene.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322231186477 – Supplemental material for SARS-CoV-2 Spike Protein Mutations in Different Variants: A Comparison Between Vaccinated and Unvaccinated Population in Western Amazonia
Supplemental material, sj-docx-1-bbi-10.1177_11779322231186477 for SARS-CoV-2 Spike Protein Mutations in Different Variants: A Comparison Between Vaccinated and Unvaccinated Population in Western Amazonia by Gabriella Sgorlon, Tárcio Peixoto Roca, Ana Maisa Passos-Silva, Márlon Grégori Flores Custódio, Jackson Alves da Silva Queiroz, André Luiz Ferreira da Silva, Karolaine Santos Teixeira, Flávia Serrano Batista, Juan Miguel Villalobos Salcedo, Rita de Cassia P. Rampazzo, Felipe Gomes Naveca and Deusilene Vieira in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
This study was developed by a group of researchers from Laboratório de Virologia Molecular da Fundação Oswaldo Cruz, in Rondônia, with financial support from the Fiocruz Genomic Network, Departamento de Ciência e Tecnologia (DECIT), Fundação para o Desenvolvimento das Ações Científicas e Tecnológicas da Pesquisa do Estado de Rondônia—FAPERO, Programa de Pesquisa para o SUS (PPSUS), and Instituto Nacional de Ciência e Tecnologia de Epidemiologia da Amazônia Ocidental—INCT EpiAmO who have been important contributors to scientific development in the Amazon region. Collaboration from Coordenação de Aperfeiçoamento Pessoal de Nível Superior—CAPES, from whom some authors received financial aid (scholarships) during the production of this study, the Vice president of Vigilância em Saúde e Laboratórios de Referências of Fiocruz, Instituto de Biologia Molecular do Paraná (IBMP), and Laboratório Central de Saúde Pública de Rondônia (LACEN/RO) were essential for the development of the study.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Fundação Oswaldo Cruz de Rondônia—FIOCRUZ/RO (PROEP 2021 Process: VPGDI-008-FIO-21-2-17), Departamento de Ciência e Tecnologia (DECIT), Fundação para o Desenvolvimento da Ação Científica e Tecnológica e à Pesquisa do Estado de Rondônia—FAPERO (Process: 01133100038-0000.72/2016; Public bid invitation: 012/2016 PRO-RONDÔNIA and PPSUS 001/2021 Process: 350.095.442.048.526.000.000), and by Instituto Nacional de Epidemiologia da Amazônia Ocidental—INCT EpiAmO. FGN is a CNPq fellow. DECIT of the Brazilian MoH, US/CDC and OPAS, Brazilian office.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
GS and DV contributed to conceptualization of this study; GS, KST, and TPR participated in data curation; GS, TPR, and DV participated in formal analysis; JMVS, FGN, and DV contributed in funding acquisition; GS performed investigation; GS, TPR, FGN, and DV participated in methodology; DV participated in project administration and supervision; GS, TPR, AMPS, JADSQ, ALFDS, and DV contributed to writing original draft; JMVS, RDCPR, FGN, and DV contributed to writing—review and editing. All authors have read and agreed to the published version of the article.
Institutional Review Board Statement
The project was evaluated and approved by the Research Ethics Committee of the Research Center for Tropical Medicine—CEPEM—Rondônia under protocol no. 4000086 and performed in accordance with the ethical principles stipulated by the 1975 World Medical Assembly and the Ministry of Health (Resolution 466).
Data Availability
All the SARS-CoV-2 genomes generated and analyzed in this study are available in the EpiCov database in GISAID under the following ID numbers: EPI_ISL_11112665-11112679, EPI_ISL_11112681-11112700, EPI_ISL_11112702-11112704, EPI_ISL_11112706-11112719, EPI_ISL_11112721-11112725, EPI_ISL_11112727-11112746, EPI_ISL_11112748-11112760, EPI_ISL_11112762, EPI_ISL_11112764-11112768, EPI_ISL_11112770, EPI_ISL_11112772-11112776, EPI_ISL_11112778-11112789, EPI_ISL_11112791-11112798, EPI_ISL_11622642-11622700, EPI_ISL_11622702-11622725, EPI_ISL_5030021, EPI_ISL_6575689-6575706, EPI_ISL_6575708, EPI_ ISL_6575710-6575739, EPI_ISL_8623163-8623164, EPI_ISL_8623166-8623256, EPI_ISL_8623258-8623269, EPI_ISL_9414682-9414748, EPI_ISL_9414750-9414774, EPI_ISL_9636793-9636797, EPI_ISL_9636798-9636804, and EPI_ISL_9636805-9636878.The list of accession IDs may be found in the attached file in the supplemental materials.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.