
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that emerged in late 2019 has accumulated a series of point mutations and evolved into several variants of concern (VOCs), some of which are more transmissible and potentially more severe than the original strain. The most notable VOCs are Alpha, Beta, Gamma, Delta, and Omicron, which have spread to various parts of the world. This study conducted surveillance in Jashore, Bangladesh to identify the prevalence of SARS-CoV-2 coinfected with dengue virus and their genomic effect on the emergence of VOCs. A hospital-based COVID-19 surveillance from June to August, 2021 identified 9 453 positive patients in the surveillance area. The study enrolled 572 randomly selected COVID-19-positive patients, of which 11 (2%) had dengue viral coinfection. Whole genome sequences of SARS-CoV-2 were analyzed and compared between coinfection positive and negative group. In addition, we extracted 185 genome sequences from GISAID to investigate the cross-correlation function between SARS-CoV-2 mutations and VOC; multiple ARIMAX(p,d,q) models were developed to estimate the average number of amino acid (aa) substitution among different SARS-CoV-2 VOCs. The results of the study showed that the coinfection group had an average of 30.6 (±1.7) aa substitutions in SARS-CoV-2, whereas the dengue-negative COVID-19 group had that average of 25.6 (±1.8; P < .01). The coinfection group showed a significant difference of aa substitutions in open reading frame (ORF) and N-protein when compared to dengue-negative group (P = .03). Our ARIMAX models estimated that the emergence of SARS-CoV-2 variants Delta required additional 9 to 12 aa substitutions than Alpha, Beta, or Gamma variant. The emergence of Omicron accumulated additional 19 (95% confidence interval [CI]: 15.74, 21.95) aa substitution than Delta. Increased number of point mutations in SARS-CoV-2 genome identified from coinfected cases could be due to the compromised immune function of host and induced adaptability of pathogens during coinfections. As a result, new variants might be emerged when series of coinfection events occur during concurrent two epidemics.
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus has been under constant observance due to its continuously emerging variants since December 2019. 1 The WHO-defined variants of concern (VOCs) until 2022 were Alpha (September 2020), Beta (September 2020), Gamma (December 2020), Delta (December 2020), Eta (December 2020), and Omicron (November 2021). 2 One of the most convincing hypotheses prevailing that the variants are originated from chronic infections in immunocompromised individuals where the virus is able to establish a persistent infection.3,4 Those VOCs showed better potential in terms of pathogenicity, virulence, and transmission rate and recent variants exhibit lower antibody neutralization sensitivity.5,6 In addition to SARS-CoV-2, other seasonal viruses such as influenza, dengue, and respiratory syncytial virus (RSV) continue to circulate in the population.7-9 Those coinfections or secondary infections are often associated with disease severity and mortality 10 although their frequency is not well known.
One study reported a high case fatality rate among COVID-19 and flu viral coinfections (r = 0.2). 7 The coinfection of dengue and COVID-19 is a matter of concern, especially in dengue-endemic regions, as it can pose serious challenge to the health system. 8 ,11-15
Although the clinical manifestations of dengue,16,17 and COVID-19 are similar in some patients, the diagnosis may not be affected by dengue virus. However, the generation of cross-reactive antibodies during dengue virus infection18,19 influence the antibody-dependent enhancement and make the cases extremely complicated.14,20,21 Analyzing the whole-genome sequences of SARS-CoV-2 among the Dengue virus coinfection cases can help in understanding the potential effects of coinfections on the evolution of SARS-CoV-2 virus. However, such studies are still in their early stages, and more research is needed to establish any link between coinfection and genomic mutations. 22 The objective of this study is to explore the whole-genome sequences of SARS-CoV-2 among the dengue virus coinfection cases, SARS-CoV-2 mutation analysis, and predict their correlation with coinfection and emerging variants.
Methods
Sample size calculation
Proportion was used to calculate the minimum sample size using the adjusted formula. 23 We have considered a proportion of the indicators as 0.5 (P = 0.5) to give a conservative estimate of the sample size, 5% two-sided statistical level of significance (Z0.05/2 = 1.96, 5% margin of error (e = 0.05) and corrected size of sample for the nonresponse as well as large population size using design effect (DEFF = 1.50) and Cochran’s corrected sample size determination formula (equation 2), respectively, which results in a sample size of 544

The adjusted sample size formula is given as

Study population
The study was conducted during the COVID-19 national surveillance at Genome Centre, Jashore University of Science and Technology (JUST), Bangladesh with the left-over samples. 10 Genome Center JUST tested a total of 27 993 samples from June 1, 2021 to August 31, 2021 and reported 9 453 (34%) COVID-19-positive cases in Jashore, Bangladesh. 24 Randomly selected 750 cases (8%, N = 9 453) were included for the verbal interview to reach the estimated sample size. The inclusion criteria in this coinfection study were SARS-CoV-2-positive cases diagnosed by Novel Coronavirus Nucleic Acid Diagnostic Kit (Sansure Biotech Inc., China) following the manufacturer’s instruction. The cases were excluded from the study if the patients are non-responsive over phone call or unable to provide the informed consent.
Spatial distribution mapping
Spatial distributions of COVID-19 cases were plotted using the ArcGIS (version 10.6) software. The approximate geolocations of the COVID-19-positive cases were pointed in Google Earth Pro platform.
Whole-genome sequencing
The SARS-CoV-2 genome sequencing was performed on samples from dengue coinfection–positive and age-, sex-, and time-matched dengue-negative cases. The sequencing procedure were performed as described elsewhere. 2 > In brief, 280 µL of nasopharyngeal samples were processed for viral RNA extraction using QIAamp Viral RNA Mini Kit according to manufacturer’s instruction. First-strand cDNAs were from the extracted viral RNA. The Ion AmpliSeq™ SARSCoV-2 Research Panel (Thermo Fisher Scientific, USA) was used to prepare libraries and target amplification. Each amplified sample was partially digested with FuPa reagents and ligated with Ion P1 adapter and Ion Xpress™ Barcode Adapters 1–16 kit (Ion Torrent™, Thermo Fisher Scientific, USA). All equimolar libraries were pooled for the preparation of template-positive Ion Sphere™ Particles (ISPs) using Ion 530™ Kit—OT2 (Thermo Fisher Scientific, USA) on the Ion One Touch™ 2 System and finally sequenced in Ion S5™ System.
Statistical analysis
Descriptive analysis was conducted from the study patients using frequency distribution to explore the sociodemographic profile, COVID-19-related experience, and information about hospitalization. The coinfection cases were observed as rare events and provided a smaller size of a sample (11 out of 572 COVID-19-positive cases). In this circumstance, this study adopted non-parametric test procedure to obtain trustworthy inferences. 25 This study employed non-parametric Chi-square test with Yates continuity correction to compare respondents’ characteristics and clinical features between COVID-19-positive and coinfected groups. In addition, the study employed non-parametric Mann-Whitney U-test to compare the mean amino acid (aa) substitution between COVID-19-positive and coinfected groups. The non-parametric Kruskal-Wallis H test were performed to identify the equality of mean mutations including pairwise variant comparison by protein change.
Time-series analysis of aa substitution
A total of 185 SARS-COV-2 whole-genome data were extracted from the GISAID database from January 2021 to October 2022. Each month we collected five to seven high-quality genome sequences mostly from Bangladeshi COVID-19 patients. We organized the genomic information based on their WHO-defined VOC and nonsynonymous mutations. We developed an ARIMA model of nonsynonymous mutations in S, open reading frame (ORF), N, M, E protein, and overall protein change applying usual procedure of identification, estimation, and diagnostics. 26 It is to be noted 27 that the number of mutations depends on the previous number of mutations as well as the previous VOC, which exhibits a nonstationary time series. This method involves successive differentiation and/or transformation to make the series stationary in order to determine the order of integration (d), as well as the development of autocorrelation and partial autocorrelation functions to determine the autoregressive (p) and moving average (q) order of the original series of SARS-CoV-2 mutation. In addition, the cross-correlation function was incorporated to identify the significant lag (l) of WHO-defined VOCs to explain the aa substitution. Let Mt and Ct be the integrated (if different) or non-integrated series of mutation and VOC respectively; thus, the ARIMAX (p, d, q) model can be written as
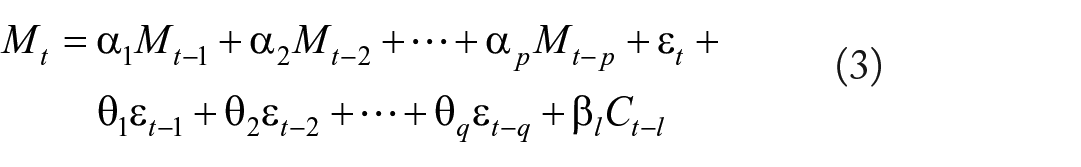
where ε t is the white noise and α, θ, and β are the parameters of AR process, MA process, and exogenous variables. 27 The Bayesian information criteria were employed to identify the optimal model with the correct combination of p, d, and q order; accordingly, the coefficient of exogenous variable estimates the average number of aa substitutions for a shift to VOC groups. It should be noted that the residual assumptions of autocorrelation, normality, and stationarity were tested after the estimation of model parameters.
Results
The prevalence of COVID-19-positive cases were 34% (9 453 out of 27 993) from June 2021 to August 2021 in Jashore, Bangladesh. This study included 750 patients out of 9 453 positive cases, of which 153 cases (20.4%) were nonresponsive in phone call and 25 personnel (3.3%) did not provide verbal consent. The remaining 572 cases provided the verbal consent for the interview and were included in the study. The study found 11 cases of dengue-COVID-19 coinfection (1.9%) out of 572 COVID-19-positive cases in the study area.
Demographic features of the patients
The male-female ratio of the COVID-19-positive patients was 271 (47.4%): 301 (52.6%). The coinfection was more common among the male patients (P = .089). Among the dengue-COVID-19 coinfection-positive cases, clinical manifestations appeared in 100% of cases compared to 87% of negative cases. Coinfected patients were more likely to experience dry cough, loss of smell and taste, and headache (P < .05). Fever appeared in 100% coinfected patients compared to 78% of the dengue-negative cases, and muscle pain was more common among the coinfected cases (45% vs 27%). There was no reported death among the coinfected cases, whereas 4 COVID-19-positive cases died out of 561 (0.7%; see Table 1).
Descriptive statistical analysis of the study-enrolled patients.

Spatial distribution of COVID-19 and dengue
Coronavirus disease 2019 was most prevalent in Avoynagar (N = 305) and Jashore Sadar (N = 85) during the study period, where the Dengue-COVID-19 coinfections rates were 2.9% (n = 9) and 2.3% (n = 2), respectively (see Figure 1).
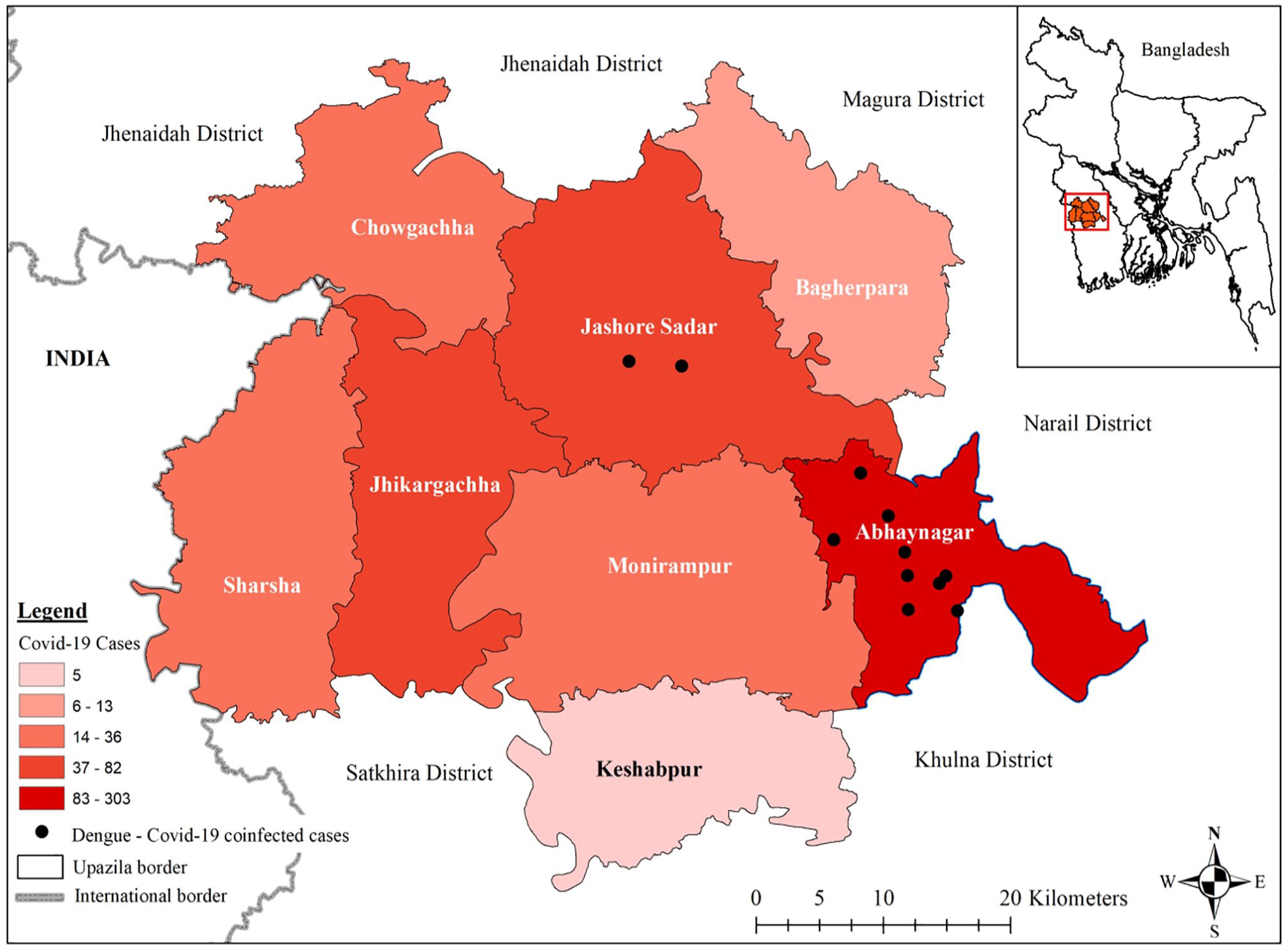
Geospatial location of Dengue-COVID-19-coinfected cases in Jashore, Bangladesh from June-August 2021.
In Vivo whole-genome sequence analysis
Seven out of 11 Dengue-COVID-19-coinfected patients and five out of nine dengue-negative COVID-19-positive patients were selected for SARS-CoV-2 genomic analysis, which provided greater than 29 500 nucleotide sequence data (see Table 2). Remaining eight samples provided only 20 000 to 26 000 genomic data and were not considered for the mutation analysis. The average number of total nucleotide substitutions among the coinfected cases were 40.9 (±2.8) compared to 34.2 (±1.7) among COVID-19 cases (P < .001). The average differences of aa substitutions were 30.6 (±1.7) among the coinfected group and 25.6 (±1.8) among the latter group. Non-parametric Mann-Whitney U-test found significant differences of aa substitution in coinfected group (P < .01). The ORF (P = .003) and N-protein (P = .03) also found significant differences between the groups (see Figure 2).
Genomic mutation analysis of Dengue-COVID-19 coinfection cases compared to COVID-19 cases.

COVID-19, coronavirus disease 2019; ORF, open reading frame.
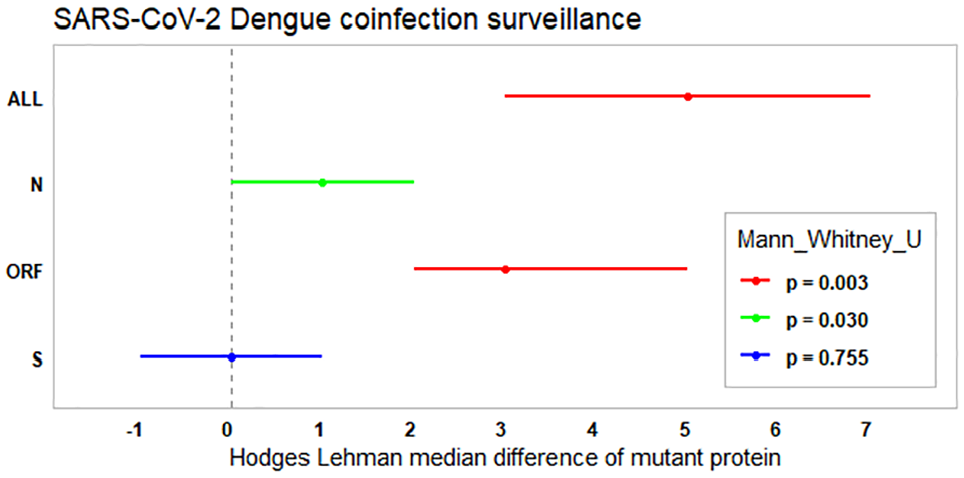
Non-parametric Mann-Whitney U-test results indicate significant differences of amino acid substitution in SARS CoV-2 proteins between Dengue-COVID-19-coinfected and dengue-negative COVID-19 patients. All samples were collected in Jashore, Bangladesh.
Protein heat map
The heat map analysis presented the changes of aa in the respective protein between coinfection-positive and -negative cases. Membrane proteins had one aa substitution, while nucleocapsid proteins had seven, non-structural proteins (ORF) had 45, and spike proteins had 15. Among the non-structural proteins, NS7a_L116 F, NSP12_P323L, NSP2_A318V, and NSP3_H1274Y were more commonly found in coinfected cases. On the contrary, NSP14_A394V, NSP3_A488S, NSP3_P1228L, NSP4_T492I, and NSP6_T77A were commonly found in dengue-negative cases. In spike proteins, few aa substitutions were observed in five strains (see Figure 3).

WGS analysis of SARS CoV-2 generated heat map of amino acid substitutions among dengue-positive and -negative groups.
In silico time-series analysis of mean aa substitution
From 185 SARS-CoV-2 genome sequence extracted from GISAID, 21 were Alpha, 20 were Beta, 21 were Gamma, 59 were Delta, 20 were Eta, and 44 were Omicron (see Figure 4). Mutation analysis revealed that aa substitutions in spike and ORF protein was increasing continuously (see Figure 5). The non-parametric Kruskal-Wallis H test results identify different pairs of VOCs in which the mean aa substitution differs significantly. The aa substitution in Alpha, Beta, Gamma, and Eta differs significantly than Delta and Omicron. Omicron variants also had significant aa substitutions (see Table 3).
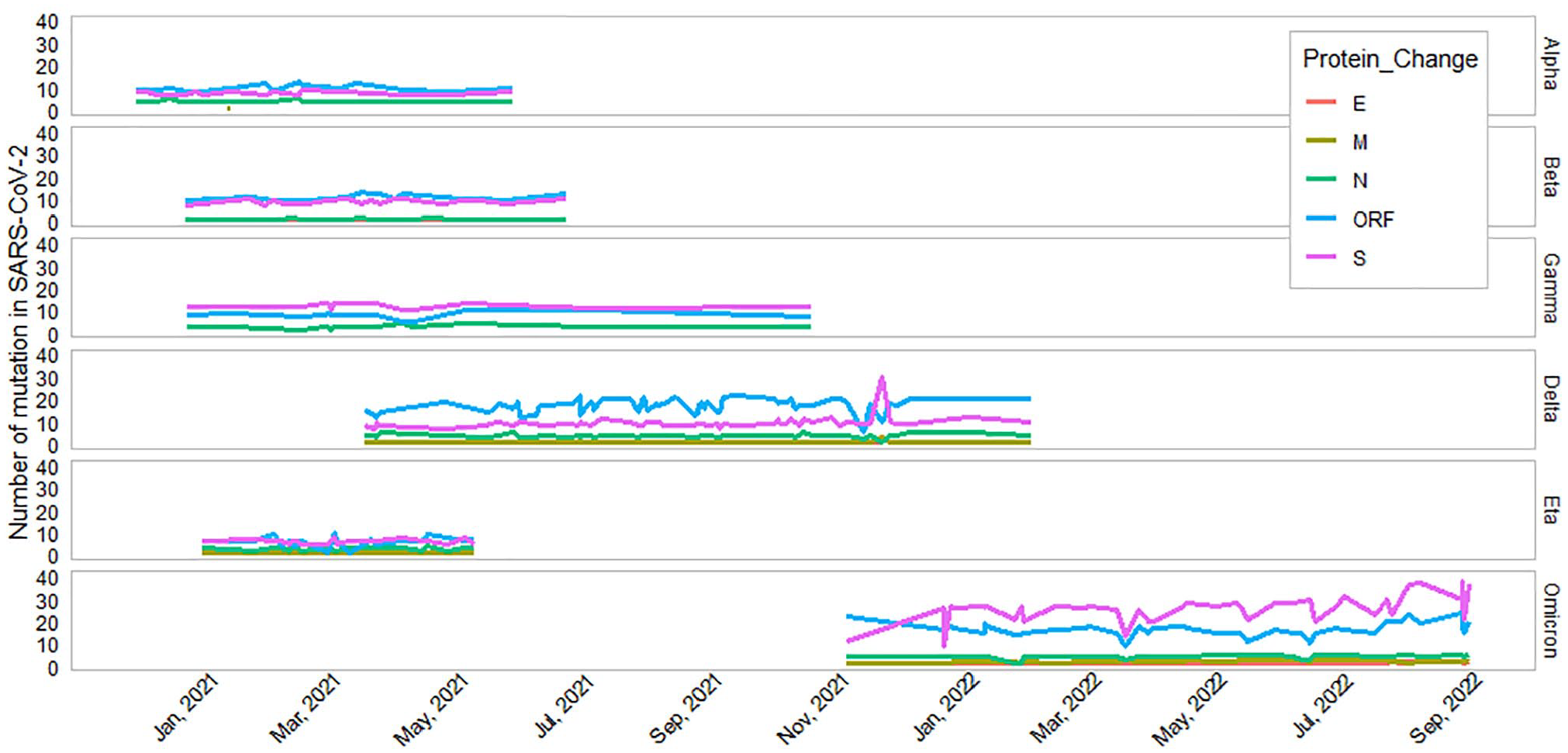
Time-series plot with frequency of amino acid substitution in WHO-defined SARS CoV-2 variants of concern (VOC) detected in Bangladesh. All SARS CoV-2 genomic data were extracted from GISAID.
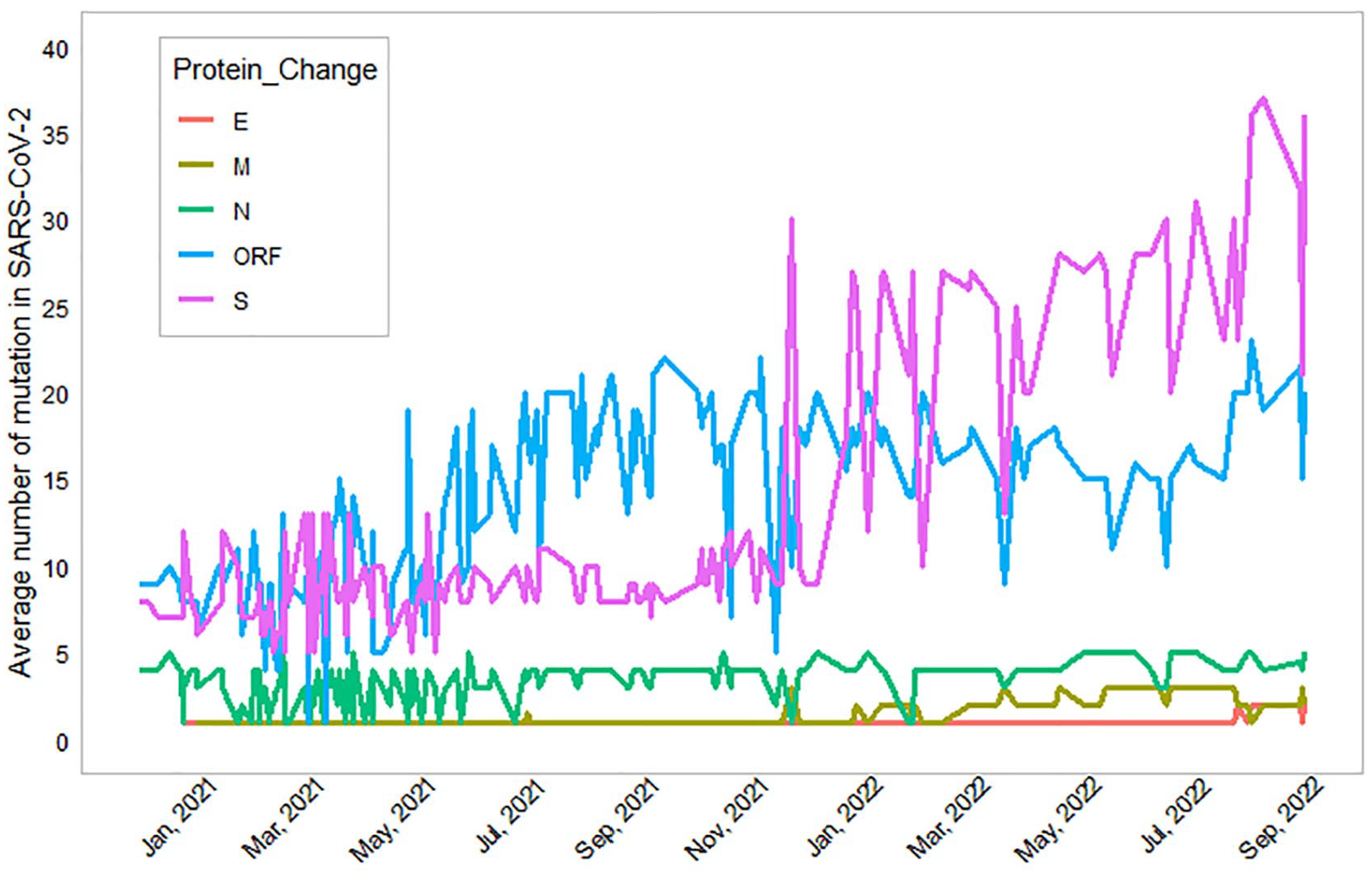
Frequency of mutations in SARS-CoV-2 protein types from January 2021 to October 2022. All SARS CoV-2 genomic data were extracted from GISAID.
Mean ± standard deviation of aa substitution in SARS-CoV-2 strains and Kruskal-Wallis H test for the equality of means including pairwise comparison by protein change.

aa, amino acid; ORF, open reading frame; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; VOC, variant of concern; WHO, World Health Organization.
Means within a column followed by different superscripted letters are statistically significantly (P < .05) different based on non-parametric pairwise comparison.
Our time-series analysis found the parameters estimates of the exogenous variables with 95% confidence interval (CI) for each aa substitution (see Table 4). According to the estimated results, emergence of SARS-CoV-2 variants from Alpha to Delta or Omicron required an average of 11.1 or 29.5 additional mutations, respectively. An average of 10.1 and 28.3 additional mutations were required from Beta variant to Delta and Omicron, respectively. From Gamma variant, Delta and Omicron accumulated an average of 8.5 and 27.0 mutations, respectively. From Delta or Eta to Omicron, 18.9 and 33.9 additional mutations took place (see Figure 6). Average mutations with 95% CI in protein category (S, ORF, N, M, and E proteins) were also mentioned in Table 4.
Estimated parameters, 95% confidence interval of the exogenous variables in the estimated ARIMAX model.

ORF, open reading frame.
All SARS CoV-2 genomic data were extracted from GISAID.
Indicates that the relationship between WHO clade category and SARS-CoV-2 genomic mutation is statistically significant at 1% and 5% level, respectively.
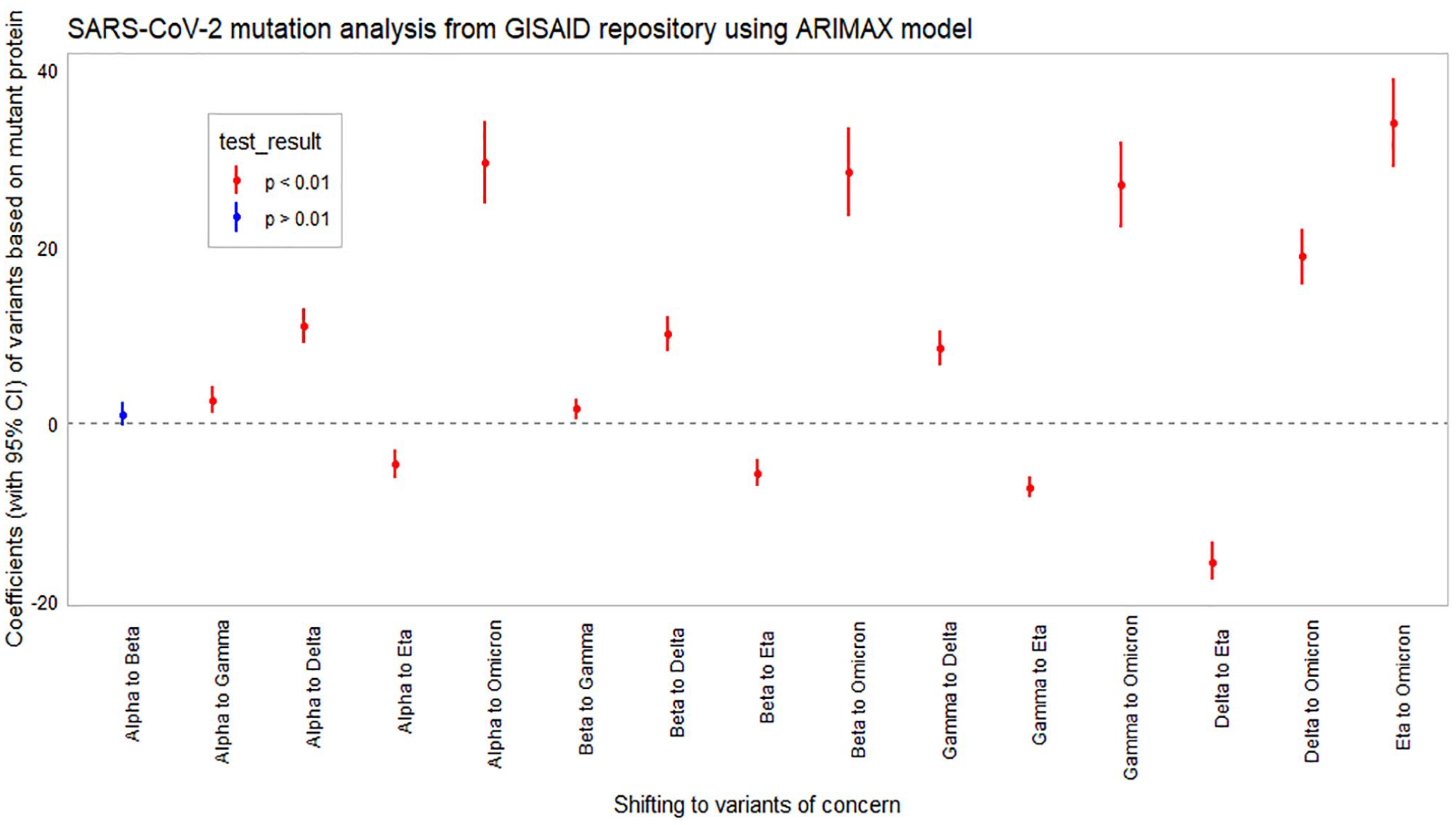
Coefficients of variants with 95% confidence interval among the different variants of concern. The differences were plotted based on non-synonymous mutations in SARS-CoV-2. Significant (P < .01) changes were highlighted in red marks.
Discussion
Dengue infection can cause a wide range of clinical manifestations, from asymptomatic or mild fever to potentially fatal diseases, such as dengue hemorrhagic fever or dengue shock syndrome. The immune response to dengue infection involves B cells, T cells, and interferons, but the virus uses various strategies to evade this response. 28 In response to dengue infection, cytokines such as interferon alpha beta (IFN-αβ), interleukin 6 (IL-6), IL-8, migration inhibitory factor (MIF), tumor necrosis factor alpha (TNF-α), IFN-γ, IL-10, and vascular endothelial growth factor (VEGF) can regulate viral replication and direct immune cells to the infection site. If this response fails, viremia develops and an amplified cytokine response leads to disease severity and affects other organs. 29 Coinfection with SARS-CoV-2 manifested severe symptoms in our study patients. We observed significant changes in clinical manifestations, specifically headache, dry cough, loss of taste and smell. The clinical outcome of the patient remained similar to some symptoms except for weakness, fever, and muscle pain (see Table 1). However, we did not perform the non-parametric chi-square test in symptoms that had low respondents. Furthermore, we identified low number of reported death cases (0% vs 0.7%) like other studies regarding the fatality in coinfected group.8,30
The presence of dengue virus coinfection with SARS-CoV-2 may have significant implications on the severity and outcome of the disease. The hypercoagulable condition resulting from high cytokine levels and ACE2 downregulation, which are typical of SARS-CoV-2, can exacerbate the interruption of the fibrinolysis system caused by dengue, potentially leading to hemoptysis. 31 The ACE2 receptors are also present in liver. 32 The impact of dengue results in fatty liver and acute liver failure. The whole spectrum of liver illness caused by dengue can progress from an asymptomatic increase in hepatic transaminases to the manifestation of abrupt liver failure. Between 4% and 52% of adult dengue victims get liver enlargement. Raised liver function results in persistent abnormality among 45% to 96% patients. 33 Furthermore, the use of ACE2 as a receptor by SARS-CoV- 2 may cause severe damage to the liver, which is already weakened by dengue. A weaker liver, elevated cytokine levels, and downregulated ACE2 all contribute to a suppressed immune system, which can prolong the duration of the virus in the body. Through its interactions with the renin-angiotensin system (RAS), ACE2 controls the immunological response.34-36 This suppressed immune system may also facilitate the longer survival and replication of other viruses, including SARS-CoV-2, leading to the emergence of new variants. This study found that the mean aa substitution in SARS-CoV-2 viruses was significantly higher among patients infected with both dengue and SARS-CoV-2 viruses compared to those who were infected with SARS-CoV-2 virus only (P < .01; see Figure 2). This finding suggests that coinfection may be paving the way for the emergence of new variants of the virus, which could have implications for the effectiveness of the current treatments. 37
There are significant differences in the aa sequences of various proteins between coinfection-positive and -negative cases. The changes were observed in the membrane, nucleocapsid, nonstructural (ORF), and spike proteins (see Table 2). The aa substitutions found between the groups exhibited some differences in several proteins such as NS7a (ORF7a), NSP1, NSP2, NSP3, NSP4, NSP6, NSP12, NSP14, and spike proteins (see Figure 3). Most of them are for replication and adaptation of the virus into a new host. 38 ,39 The SARS-CoV-2 ORF7a has immunomodulatory function and it can counteract the antiviral effect the host cell has on the budding virions. The NS7a_L116 F mutation resides in the end-loop of the C-terminal domain. The “indel” mutation in ORF7a usually causes destabilization of protein structure and thus can enhance hindrance from human immune response. 40 This particular mutation seen in our study sequences might be able to affect viral replication and thus can lead to antigenic drift. 41 The NSP2_A318V mutation was reported in Malaysia and other countries, but no specific effect of the mutations has been reported yet.42-44 The NSP3_H1274Y mutation was reported in Pakistan 44 and the Australian Capital Territory (ACT) region at high levels. 45 This mutation resides in another important deletion mutation (S1265-(Serine→deletion) and L1266I (Leucine→Isoleucine) in the βSM region) reported in a recent study. 46 This disordered βSM region has, however, no definite functional role. The NSP12_P323L, coevolving D614G and P323L mutations in SARS-CoV-2 are associated with the severity of COVID-19, 47 infectivity and survival ability of the viral strain. 48
This study has a potential limitation where the prevalence (1.9%) of coinfection may have been underestimated due to only clinically suspected cases being recommended for the dengue virus testing with NS1 antigen and IgG antibodies. Moreover, we did not consider any case as coinfection positive if any patient was unable to show both positive test records. It is also important to note that a large number of selected cases were nonresponsive in phone calls and did not provide the consent, which may introduce some bias into the sample (see Figure 1).
The study used an ARIMAX model from the GISAID repository to estimate effect of point mutations in SARS-CoV-2 variants over time. We designed the model in each protein change by keeping each VOC category as a reference to avoid multi-collinearity. The order of ARIMAX model with minimum Bayesian information criteria including smaller residual variance from the indication of autocorrelation and partial autocorrelation function of the stationary series of mutation (see Table 5). ARIMAX (2,1,0) model in S-Protein for Alpha to Omicron indicates that the later variant might be evolved in the time of lag 2; more specifically, previous two time points. ARIMAX (0,1,1) indicates that the mutation is related with the errors of lag 1 and ARIMAX (0,0,0) indicates the white noise process (see Table 5). However, we were interested to estimate the coefficient of exogenous variables using proper ARIMAX model instead of forecasting out-of-sample mutations. The negative coefficient with significant effect of corresponding exogenous variables indicated that Eta did not emerge from Alpha, Beta, Gamma, or Delta variants. It could be generated from the wild-type variant.
The autoregressive (p) and moving average (q) order of the integrated (d) or non-integrated (d = 0) series of mutation for each protein change with reference of WHO-defined variants of concern category.

ORF, open reading frame.
The in silico analysis of SARS-CoV-2 sequences found that the frequency of aa substitution in Spike and ORF proteins increased over time (see Figure 4). Our time-series plot indicated that the emergence of new VOC was mostly due to the mutations in spike protein and ORF protein (see Figure 5). The study also found that the Delta variants mostly emerged from changes in ORF proteins, while the Omicron variants emerged with changes in both Spike and ORF proteins. Our dengue and COVID-19 coinfection study was placed from June to August of 2021 when the Delta variant was predominant in Bangladesh. The study estimated that the SARS-CoV-2 Omicron variant accumulated an average of 18.9 (95% CI: 15.74, 21.95) non-synonymous mutations than Delta. Our hospital-based surveillance observed that one coinfection cycle caused five non-synonymous mutations on top (mean 25.6 vs 30.6) and suggested that existing COVID-19 pandemics might come up with new variants of concern soon. 49 As for the potential emergence of new variants at the end of the dengue season, it is indeed possible that the seasonal spread of dengue could infect along with the SARS-CoV-2 and evolve as a new variant. One of our studies is reporting that the 2023 dengue outbreak had a higher morbidity rate compared to previous years (2019-2022) in Southwest of Bangladesh. 50 That outbreak was caused by DENV-2 Cosmopolitan genotype that was evolved as a new variant among C clade. The outbreaks of 2017 and 2018 in Bangladesh were also caused by DENV-2 Cosmopolitan genotype within B and C clade, 51 although the 2023 variant did not resemble any previous Bangladeshi C clade variants (2017, 2018, and 2019), suggesting a shift of variants.
Conclusions
The SARS-CoV-2, despite having proof-reading activity, continues to produce new variants since 2020. The RdRp of SARS-CoV-2 can correct errors during replication, which contribute to the stability of the virus genome. Nonetheless, the emergence of new variants took place due to its high rate of replication and mutation of the virus, combined with selective pressure from the host immune responses and coinfection with other viruses. Although there was no recombination event evident in our study, our mutation analysis found a significant difference in the frequency of point mutations among the coinfected cases. The findings of our study noted that coinfection with two different viruses can potentially lead to the emergence of new variants. The emergence of new variants and their potential impact on the ongoing pandemic further highlights the need for research on coinfection and their potential effects on viral evolution and pathogenicity. Continuous monitoring and study of the evolution of the virus and its variants is required to predict and prepare for potential future outbreaks. This can help public health officials and policymakers develop effective measures to control the spread of the virus, including developing new vaccines and treatments.
Footnotes
Acknowledgements
We would like to acknowledge Genome center, Jashore University of Science and Technology and the Ministry of DG Health, Bangladesh Government.
Author Contributions
HMA contributed toward conceptualization, methodology, validation, formal analysis, investigation, visualization, writing original draft, funding acquisition, and project administration. FR contributed to software, formal analysis, and data curation. LS, PM, SY, PS, TA, RP, and MR were involved in the investigation. PKD was involved in the investigation, resources, validation, and formal analysis. MSH was involved in investigation, resources, and validation. MSR contributed to formal analysis and data curation. SLS contributed to investigation, resources, and validation. ASMRUA contributed to writing review and editing, resources. AH, OKI, MTI, SN, and SA contributed to writing review and editing. MMR contributed to writing review and editing, resources, validation, and formal analysis. IKJ contributed to formal analysis, validation, writing review and editing, resources, supervision, and project administration. MAH contributed to writing review and editing, supervision.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was funded (JUST/Research Cell/Research Project/2020-21/FOET11) by Jashore University of Science and Technology, Jashore-7408, Bangladesh. This funding does not include any personal cost or salary.
Ethical Approval
The study has gone through the Ethical Review Board of Jashore University of Science and Technology (ERC no: ERC/FBST/JUST/2021-62) and approved the study. The participants have been informed about the purpose and the objectives of this study. Verbal consent was obtained from optimistic respondents before they were enrolled in this study, and written informed consent has been taken before the processing of the samples. We ensured that all ethical considerations have been taken into account while conducting the research involving human participants. This includes obtaining the informed consent, protecting participants’ privacy and confidentiality, and ensuring that the study was conducted in an ethical and responsible manner. The research was conducted in accordance with the applicable laws and regulation, and that any potential risks or ethical concerns were addressed and managed appropriately.
Data Availability Statement
The SARS-CoV-2 whole-genome sequences data used in this coinfection study were deposited in the GISAID database (accession no. EPI_ISL_17226262, EPI_ISL_17226263, EPI_ISL_17226264, EPI_ISL_17226265, EPI_ISL_17226266, EPI_ISL_17226267, EPI_ISL_17226268, EPI_ISL_17226269, EPI_ISL_17226270, EPI_ISL_17226271, EPI_ISL_17226272, and EPI_ISL_17229387).