
Abstract
Dengue and Japanese encephalitis virus (JEV) are mosquito-borne RNA viruses that can cause severe illness leading to death in the tropics and subtropics. Both of these viruses interact directly with the C-type lectin domain family 5, member A receptor (CLEC5A) on human macrophages which stimulates the release of proinflammatory cytokines. Since blockade of this interaction has been shown to suppress the secretion of cytokines, CLEC5A is considered a potential target for the development of new treatments to reduce virus-induced brain damage. Developing a vaccine against dengue is challenging because this virus can cause disease through 4 different serotypes. Therefore, the vaccine must immunize against all 4 serotypes to be effective, while unvaccinated people still contract JEV and suffer from its complications. Small interfering RNAs (siRNAs) play an important role in regulating gene expression by causing the degradation of target mRNAs. In this study, we attempted to rationally design potential siRNA molecules using various software, targeting the CLEC5A gene. In total, 3 siRNAs were found to be potential candidates for CLEC5A silencing. They showed good target accessibility, optimum guanine-cytosine (GC) content, the least chance of off-target effects, positive energy of folding, and strong interaction with Argonaute2 protein as denoted by a negative docking energy score. In addition, molecular dynamics simulation of the siRNA-Ago2-docked complexes showed the stability of the complexes over 1.5 nanoseconds. These predicted siRNAs might effectively downregulate the expression of the CLEC5A receptor and thus prove vital in the treatment of dengue and JEV infections.
Introduction
The dengue virus (DV), which is becoming a major source of illness and death in tropical and subtropical countries, is now a threat to approximately half of the world’s population. 1 This single-stranded RNA virus belongs to the Flaviviridae family 2 and is transmitted to humans by Aedes species mosquitoes. Each year, as many as 400 million people get infected with DV, of which around 100 million show symptoms and yet there is no specific therapy or vaccine available to prevent the infection. 3 Developing a vaccine against the virus is challenging since with 4 different serotypes of the virus that can cause the disease, the vaccine must immunize against all 4 types to be effective. 4 In addition, primary infection by a particular serotype provides lifelong immunity only against the infecting serotype and not the other ones. More importantly, secondary infections are associated with an increased risk of developing dengue hemorrhagic fever. It was discovered by Chen et al 5 that CLEC5A (C-type lectin domain family 5, member A; also known as myeloid DAP12-associating lectin [MDL-1] receptor on monocytes and macrophages) interacts directly with the dengue virion, acting as a signaling receptor for the release of cytokines. The pathogenesis of dengue infection involves massive secretion of proinflammatory cytokines from macrophages. They also showed that the knockdown of the CLEC5A gene alone was sufficient to suppress the release of cytokines from DV-infected macrophages. Thereby it was recognized that CLEC5A silencing could be a promising strategy to avoid the fatal consequences of dengue hemorrhagic fever and dengue shock syndrome. 5
The same researchers discovered in a second investigation that CLEC5A interacts with the Japanese encephalitis virus (JEV), a flavivirus that is spread by mosquitoes and is thought to be the main cause of viral encephalitis in Asia, where there are an estimated 68 000 clinical cases per year.6,7 Although vaccines are available to prevent JEV, unvaccinated individuals are at risk of contracting the infection. During JEV infection, central nervous system (CNS) inflammation is induced which involves neuronal cell death, inhibition of the proliferation and differentiation of neural progenitors, and disruption of the blood-brain barrier (BBB). Anti-CLEC5A antibodies have been demonstrated to protect JEV-infected mice against BBB disruption and limit the infiltration of inflammatory myeloid cells into the brain. This is because the CLEC5A receptor interacts with JEV to activate monocytes and macrophages. 7 It is critical to establish an efficient curative therapy for JE patients because the case-fatality rate among those with encephalitis can be as high as 30%, and 30% to 50% of those who survive suffer long-term neurological sequelae. 8 CLEC5A can be considered a potential target to reduce virus-induced brain damage. 7
Furthermore, a study in 2010 marked CLEC5A as a key regulator of synovial injury and bone erosion during autoimmune joint inflammation. It has been demonstrated that activation of CLEC5A increases the migration of inflammatory neutrophils and macrophages to the joint and encourages bone disintegration, whereas functional inhibition of the receptor decreases the clinical manifestations of autoimmune joint inflammation. These findings suggest that CLEC5A may also be a therapeutic target for the treatment of immune-mediated skeletal disorders. 9
In 2016, it was also revealed that the CLEC5A receptor interacts with the hemagglutinin protein of influenza viruses and plays once again an important role in the pathogenesis of the virus by enhancing the inflammatory response in myeloid cells. This observation made the receptor a potential therapeutic target against influenza infections as well. 10
RNA interference (RNAi) is an evolutionarily conserved naturally occurring eukaryotic process by which double-stranded RNA-like siRNA (small interfering RNA) and miRNA (microRNA) trigger posttranscriptional gene silencing. 11 Small interfering RNA is 20 to 25 base pairs in length and functions by causing mRNA to be broken down after transcription 12 resulting in no translation. Using nanoparticles like polyethyleneimine (PEI) and other methods, RNAi can be introduced into the cell for the knockdown of a gene of interest 13 and thus may be used for therapeutic purposes like chemical drugs. 14 The gene silencing efficiency of PEI nanoparticles is similar to the commercially available transfecting agent lipofectin but with reduced cytotoxicity. 15 Besides, such nonviral vectors are very effective and safe for targeting diverse tissues in vivo. Since CLEC5A is a human receptor, its expression is only to be transiently silenced as it can be sometimes beneficial to the host in fighting bacterial infections. Therefore, siRNA is preferred to shRNAs (short hairpin RNAs), another regulatory RNA molecule, as siRNAs are transiently expressed in cells, while shRNAs get stably integrated leading to long-term knockdown of the target gene. 16
Therefore, in this study, we set on to rationally design potential siRNA molecules for CLEC5A gene silencing. For accomplishing the task of designing potential RNAi candidates, multiple computational web-based tools such as similarity search, secondary structure prediction, RNA interaction evaluation, thermodynamics analysis, molecular docking, and molecular dynamics (MD) simulation were conducted. After initial prediction followed by rigorous filtration and authentication, we finally designed the 3 most potential siRNAs against the CLEC5A receptor which should transiently prevent the translation of the receptor and thus protect against the fatal symptoms of dengue and JEV infections.
Materials and Methods
CLEC5A sequence retrieval and prediction of siRNA
The coding sequence of the human CLEC5A receptor was retrieved from the National Center for Biotechnology Information (NCBI) protein portal (http://www.ncbi.nlm.nih.gov/). The entire sequence was used for designing functional and target-specific siRNAs, using the siDirect 2.0 web server (http://siDirect2.RNAi.jp/) which is an updated version of the web-based software siDirect. 17 The study of Naito et al 17 has concluded that when the candidate functional siRNAs could form seed-target duplexes with Tm values below 21.5°C, and their 19-nt regions spanning positions 2 to 20 of both strands have at least 2 mismatches to any other nontargeted transcripts, siDirect 2.0 can design at least one qualified siRNA for >94% of human mRNA sequences in RefSeq. Therefore, the maximum melting temperature (Tm value) for the formation of the seed duplex was kept below 21.5°C (the default) for reducing off-target effects and guanine-cytosine (GC) content was set between 30% and 64%. 18 In the server, 3 rules including Ui-Tei, 19 Amarzguioui, 20 and Reynolds 21 based algorithms were selected as parameters for siRNA selection (Table 1).
Algorithms/rules applied in siDirect 2.0 server for rational designing of siRNA molecules.

Abbreviation: siRNA, small interfering RNA.
The predicted siRNAs were cross-validated by the i-Score (inhibitory-Score) Designer server 22 in which siRNA prediction scores were calculated from 9 different algorithms including both first-generation algorithms and second-generation algorithms. The second-generation algorithms include i-Score, 22 s-Biopredsi, 23 and DSIR 24 which are considered more reliable than the other algorithms. In this server, the siRNAs predicted from siDirect were used as input and those which ranked in the top 10 predictions made by any of the following algorithms (s-Biopredsi, i-Score, and DSIR) in i-Score Designer analysis, were selected for further analysis. In this server, the GC content of individual siRNAs was also given as part of the prediction results. Afterwards, the GC content of each of the candidate siRNAs was calculated in OligoCalc. 25
Off-target similarity search and filtering siRNAs with palindromic sequence
Both sense and antisense strands of the siRNAs should avoid any secondary structures like palindrome that can be prohibitive to the formation of the duplex properly. 26 Therefore, the predicted siRNA sequences were screened for palindromic sequences using the EMBOSS palindrome online tool (www.bioinformatics.nl/cgi-bin/emboss/palindrome) in which the minimum length of the palindrome was set at 4 nucleotides. Next, both strands of the siRNAs were checked via blastN at the NCBI database against the Refseq-RNA database of humans to reduce the risk of silencing unintended genes. While the parameters were adjusted for short-input sequences, word size was set at 7, and the expected threshold was set at 100 to cover random matches as well.
The guide and passenger strands of the siRNAs were also screened by the Genome-wide Enrichment of Seed Sequence matches (GESS) online tool (www.flyrnai.org/gess/) 27 to further check for any potential off-target effects. The seed regions of the siRNAs were analyzed to identify potential off-targeted transcripts that could produce false-positive results. The input file contained only active RNAi reagents, the region was selected 3ʹ UTR and the reference database was the built-in human database. In a final step, whether the siRNAs contained any immunostimulatory motifs including “GUCCUUCAA” 28 and “UGUGU” 29 that could result in toxicities were also checked.
Secondary structure prediction and thermodynamics study of the siRNA-target gene interaction
The function of a regulatory RNA molecule is determined by its structure mainly. RNA tertiary interactions involve secondary structure elements and are substantially weaker than secondary interactions. Thus, to a large extent, the free energies in the secondary structure represent the thermodynamics of RNA folding. 30 In this study, the secondary structures of the guide strand of the siRNAs and their target site as well as their free energy of folding were determined using the MaxExpect program 31 of the RNA structure web server (https://rna.urmc.rochester.edu/RNA). Here, the RNAi structures with higher values of energy were considered better candidates as those molecules had less chance of folding.
The thermodynamics of RNA-RNA interaction is another major parameter for siRNA efficiency where stronger interaction denoted by lower binding energy indicates better efficacy. For this, the interaction between the guide strand and the target sequence was analyzed by the DuplexFold program of the RNAstructure web server in which 2 RNA sequences are folded into their lowest hybrid free energy conformation along with their free energy of binding being calculated. Afterwards heat capacity and concentration plots for the predicted siRNAs were generated by the DINAMelt application (http://mfold.rna.albany.edu/?q=DINAMelt/Hybrid2). 32 In the heat capacity plot, the collective heat capacity of the RNA duplex (Cp) is plotted as a function of temperature, and the melting temperature, Tm (Cp) is denoted by the local maximum of the heat capacity curve. The contributions of each species to the ensemble heat capacity are shown in another detailed plot. On the contrary, in the concentration plot, the relative concentration of 5 species (1 heterodimer, 2 homodimers, and 2 single strands) is plotted as a function of temperature, and Tm (conc) indicates the point at which the concentration of a double-stranded molecule is one-half of its maximum value.
Prediction of siRNA target site accessibility within target mRNA
The siRNA application module of S-fold (https://sfold.wadsworth.org/cgi-bin/sirna.pl) was applied to determine the target site accessibility of the predicted siRNA molecules. The target sequences of the siRNAs were given as input and the accessibility of target sites within the target mRNA for specific siRNAs was then assessed by the server through multiple parameters. Several siRNA parameters are being provided by S-fold, including target site accessibility score, differential stability of siRNA duplex ends (DSSE), average internal stability at the cleavage site (AIS), total stability of the siRNA duplex, and so on.
Modeling and docking of argonaute 2 protein and the siRNAs
The 3-dimensional structure of argonaute 2 (Ago2) protein was predicted using SWISS-MODEL (https://swissmodel.expasy.org/interactive) 33 based on the crystal structure of RNA-bound human AGO2 protein (Protein Data Bank [PDB] identifier: 4Z4D) via homology modeling. The model was further refined by the GalaxyRefine (http://galaxy.seoklab.org/refine) server 34 which can improve structure quality beyond the scope of template information, through MD simulation. Next, the quality of the models was checked using ERRAT 35 based on the quality scores from ERRAT and GalaxyRefine, and one of the refined models was finally selected.
On the contrary, the 3D structure of the guide RNA was modeled in BIOVIA Discovery Studio 2021 in which the RNA structure bound to the Ago2 protein (PDB identifier: 4Z4D) was taken as a template. The sequence of the template guide RNA was mutated according to the predicted siRNA sequence so that similar conformation could be retained. Afterwards, the geometry of the siRNAs was optimized in Avogadro software 36 using Merck molecular force field 94 under the steepest descent algorithm for 1000 steps and 10e−7 convergence.
Finally, molecular docking between the Ago2 and the siRNAs was performed in the HDOCK server (http://hdock.phys.hust.edu.cn/) which is a hybrid docking algorithm employing both free docking and template-based modeling with high efficacy and robustness. 37 The resulting complexes with the lowest docking energy scores and closest siRNA binding position to the template were finally selected and visualized for further analysis.
MD simulation of siRNA (guide strand)—Ago2 protein docked complex
Protein complexes can be dynamic in nature as they interconvert between several alternate configurations. Therefore, MD simulation is considered a reliable tool for studying the stability of macromolecular complexes, especially how biomolecules will respond upon the addition or removal of a ligand. 38 In this study, MD simulation of the siRNA-Ago2 docked complexes was performed with the help of the NAMD 2.14 package. 39 For this, the input files were generated using the CHARMM GUI server 40 where the docked protein complexes were solvated and NaCl ions were added to the simulation box. A periodic boundary condition was incorporated to perform the simulation which was retrieved using VMD 1.9.3 software. 41 In this work, the thermodynamic parameter was set at 310 K. NAMD 2.14 software calculates frame step trajectory files (each frame step corresponds to every time step). Finally, the simulations were evaluated through root mean square deviation (RMSD) and root mean square fluctuation (RMSF) analysis through R package Bio3D developed by Grant laboratory.
The workflow of the entire study is depicted in Figure 1.
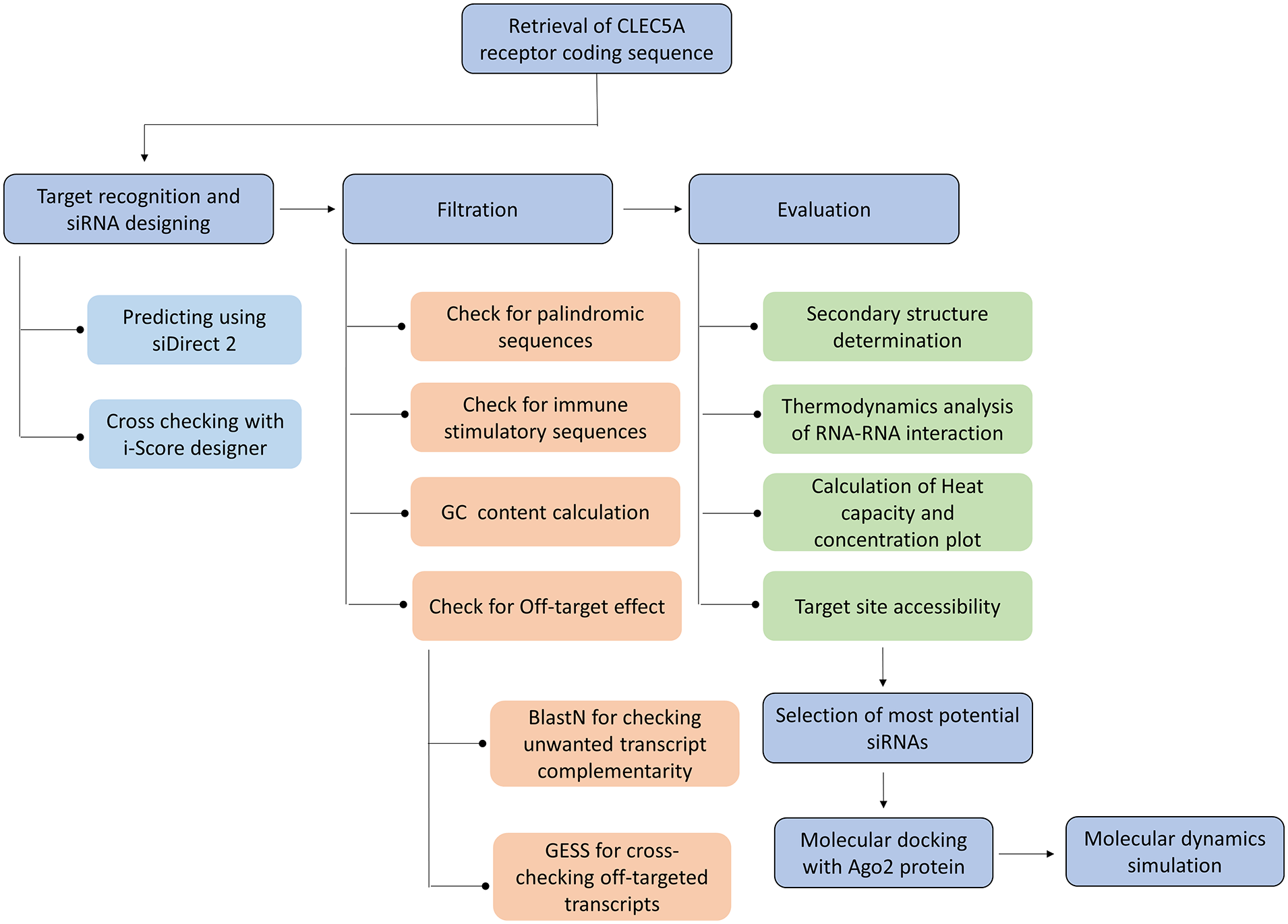
Graphical representation of the workflow used in this study. CLEC5A indicates C-type lectin domain family 5, member A receptor; GESS, genome-wide enrichment of seed sequence matches; siRNA, small interfering RNA.
Results
siDirect 2.0 and i-score designer predicts siRNAs from the retrieved CLEC5A target sequence
The coding sequence of the human CLEC5A receptor was retrieved from the NCBI-Genbank database, and it consists of 567 nucleotides. siDirect 2.0 predicted in total 29 siRNAs based on the algorithms of Ui-Tei, Reynolds, and Amarzguioui as well as seed-duplex stability. Out of these 29 SiRNAs, 14 followed all 3 rules (URA). However, 10 siRNAs were finally selected for further analysis as they had optimum GC content in the range of 30% to 64% for siRNA functionality. 42 To cross-check, the predicted siRNAs were also analyzed by i-Score Designer, and 6 of them were found to rank among the top 10 predictions made by s-Biopredsi, i-Score, or DSIR prediction tools and so these siRNAs (Table 2) were selected for further filtration.
siDirect and i-Score designer predicted overlapped siRNAs along with their different parameters.

Abbreviation: siRNA, small interfering RNA.
Filtration of siRNAs for the presence of palindromes, off-target effects, and immunostimulatory action results in 3 potential siRNAs
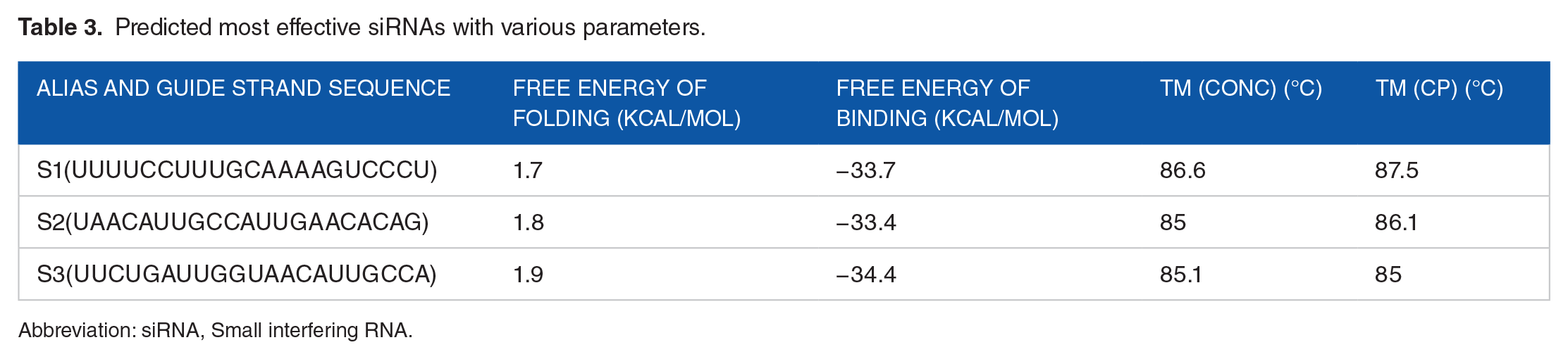
Four out of the 6 siRNAs did not have any palindromic sequences in their guide and passenger strands. These 4 siRNAs were blasted against the reference RNA database of humans, and 3 of them showed less than 78% query coverage with other genes which are considered tolerable. For further confirming this observation, when these RNAs were analyzed by GESS, the results showed that the off-targets were not significant by any of the tests including Fisher, Bonferroni, Bonferroni step-down (Holm) correction, and Benjamini and Hochberg methods. It suggests that these siRNAs would not create undesirable silencing of any other genes. In addition, the absence of the immunostimulatory motifs within these 3 siRNAs showed that the RNAs would be associated with minimal immune activation. Therefore, these 3 siRNAs were finally selected as potentially most effective siRNAs which were then subjected to further analyses (Table 3).
Predicted most effective siRNAs with various parameters.
Abbreviation: siRNA, Small interfering RNA.
Secondary structure and thermodynamics study of the candidate siRNAs
The predicted secondary structures of the guide siRNA strands are presented in Figure 2. Their free energy of folding ranged between 1.7 to 1.9 kcal/mol (Table 3). All of the candidate siRNAs displayed promising outcomes, which is consistent with the theory that siRNAs with positive free energies of folding may encourage increased permissibility to the target, improving gene silencing. On the contrary, the binding between the siRNAs and their target gene was also analyzed (Figure 3), and the resulting binding energy ranged between −33.4 and −34.4 kcal/mol (Table 3).

Secondary structures of the guide strands and target sequences of S1, S2, and S3 siRNAs along with their free energy of folding: (A) secondary structure of the guide strand of S1, (B) secondary structure of the guide strand of S2, (C) secondary structure of the guide strand of S3, (D) secondary structure of the target sequence of S1, (E) secondary structure of the target sequence of S2, and (F) secondary structure of the target sequence of S3. siRNA indicates small interfering RNA.

Structure of binding of siRNA (guide strand) and target RNA along with their predicted free energy of binding: (A) structure of interaction between guide strand and target sequence for S1 siRNA, (B) structure of interaction between guide strand and target sequence for S2 siRNA, and (C) structure of interaction between guide strand and target sequence for S3 siRNA. siRNA indicates small interfering RNA.
Using the DINAMelt web server, 32 the heat capacity plot and concentration plot were also calculated for the RNAi molecules. As temperature increases, the heat capacity of the siRNAs also increases, and the temperature at which the collective heat capacity (Cp) reaches its maximum value is denoted as the melting temperature, Tm (Cp). In the concentration plot, the concentration of the double-stranded molecule decreases with increasing temperature, and the concentration of folded and unfolded single strands increases with increasing temperature. The temperature at which the concentration of the double-stranded molecule is half of its maximum value is denoted as Tm (Conc). The higher values of these 2 melting temperatures can be considered as an indication of higher efficacy of the siRNAs. From Table 3, it can be seen that among the 3 siRNAs, S1 siRNA showed the highest Tm (Cp) and Tm (Conc) values (87.5°C and 86.6°C, respectively).
Target site accessibility and further RNA duplex thermodynamics study of the siRNAs
The parameters obtained from Sfold output for the 3 finally selected siRNAs are given in Supplementary Table 1. Through multiple experimental approaches, the significance of target accessibility for the function of siRNAs has been proven. 43 In this study from Sfold analyses, it was found that S1 siRNA had a target accessibility score of 7, whereas S2 and S3 both had a target accessibility score of 8 (Supplementary Table 1). Furthermore, according to the target accessibility rule, antisense siRNA binding energy should be less than or equal to −10 kcal/mol, and from the results, it can be seen that all of the 3 siRNAs fulfilled this criterion ranging between −14.5 and −20.2 kcal/mol.
It has also been stated that functional siRNA duplexes tend to have lower stability at the cleavage site as well as on the 5ʹ-antisense end than on the 5ʹ-sense end and the relative stability of the 2 ends influences the degree to which each strand participates in the gene silencing pathway.44,45 This difference is shown by the DSSE value (differential stability of siRNA duplex ends, in kcal/mol) which should be more than 0 kcal/mol as the guide strand gets assembled with RNA-induced silencing complex (RISC). The results demonstrate that the values for the proposed siRNAs ranged between 1 and 6 kcal/mol (Supplementary Table 1), demonstrating that ineffective and undesirable mRNA targeting that can occur from the assembly of passenger strands with RISC is avoided. Another score which is siRNA duplex feature score, was found to be 7 for all the siRNAs which can be as low as −2 and as high as 10 and values greater than 6 are acceptable.
Results from Sfold also showed that the AIS values (AIS at the cleavage site) for all the predicted siRNAs were around −6.0 kcal/mol and according to cleavage site instability rule this value should be greater than −8.6 kcal/mol. Average internal stability computes the average internal energy for positions 9 to 14 in the antisense strand (counting from 5ʹ end of antisense strand). In addition, the duplex thermodynamics score of the 3 siRNAs was found to be 2 in which 1 point was attributed to their DSSE values being greater than 0 kcal/mol, and another point was due to their AIS values being greater than −8.6 kcal/mol. Finally, the total score for these siRNA duplexes were found to be either 16 or 17 which surpassed the Sfold-imposed threshold of 12. This score sums up target accessibility score, duplex feature score, and duplex thermodynamics score and its maximum value can be 20. It can be seen that all Sfold filtration criteria have been fulfilled by these 3 siRNAs.
Molecular docking between Ago-2 protein and the siRNAs
The predicted 3D structure of Ago2 protein was refined by GalaxyRefine server which generated 5 models and calculated multiple parameters for each of the model through which the quality of the models could be assessed including MolProbity score, clash score, poor rotamers, Ramachandran favored region, and so on. In addition, the models were checked by ERRAT. All the results are shown in Supplementary Table 2.
Clash score represents the number of atom pairs in a protein model that are unusually close to each other and may result in steric clashes and therefore lower numbers are considered better. Poor rotamers indicate the number of amino acids in the protein with poor side chain geometry and a high percentage of poor rotamers (>1.5%) can be concerning. Another important factor is Ramachandran favored region within the protein which refers to the percentage of amino acids having favored phi/psi angles in the protein backbone. Ideally, this number should be higher than 98%. The next value is MolProbity score which combines all scores regarding atom contacts, geometry, and conformation into a single value to validate structure quality and once again comparatively lower values are desired. Finally, the models were also run through ERRAT program which determines whether the distribution of nonbonded atomic interactions in a candidate structure correlates with the distribution established from a database of reliable, high-resolution structures. Thus, ERRAT can be considered an overall quality factor for nonbonded interactions, with higher scores suggesting higher quality. From the results shown in Supplementary Table 2, it can be seen that model 1 had highest ERRAT score (95.45%), closest MolProbity score (1.28) to the original model and lowest clash score (11.7) among the 5 models. In addition, it showed less than 1.5% poor rotamers (0.1) and greater than 98% residues in Ramachandran favored region. Therefore, model 1 of Ago2 protein structure was selected for docking with the candidate siRNAs. The structure is given in Supplementary Figure 1.
The docked complexes between Ago2 protein and our candidate siRNAs were screened to find out in which complexes the siRNA was placed inside the PAZ and MID domains of the Ago2 protein thus maintaining a similar conformation to the PDB 4z4d structure. It was found that for S1 and S3 siRNAs, only one docked complex of each docking analysis showed the involvement of PAZ and MID domains with the siRNAs. On the contrary, for S2 siRNA 2 docked complexes had similar conformation to the 4z4d structure and based on the docking energy score, the complex with the lower docking energy was selected. The docking score, interaction statistics and interactive residues of each of the siRNA docked complexes are presented in Table 4 and the docked complexes are shown in Figure 4.
HDOCK docking energy score, number of different types of bonds within each docked complex and interactive residues of Ago2 protein in the docked complexes.

Abbreviation: siRNA, small interfering RNA.

Docked structures of S1, S2, and S3 siRNAs (cartoon view) with Ago2 proteins (surface view). (A) S1 siRNA docked with Ago2 protein (B) S2 siRNA docked with Ago2 protein and (C) S3 siRNA docked with Ago2 protein. The PIWI/Argonaute/Zwille (PAZ) domain and the Middle (MID) domain are colored as green and orange spheres, respectively, and the rest of the protein is denoted by blue color ribbon structure. The siRNAs are represented by yellow arrow structure. siRNA indicates small interfering RNA.
MD simulation of the siRNA-Ago2 docked complexes
Among the 3 predicted siRNAs, S1 and S2 showed stronger binding interaction with the Ago2 protein compared with S3, although S2 had less hydrogen bond interactions compared to S3 siRNA. S1 and S2 docked complexes were further examined through MD simulations. A 1.5 ns simulation was conducted to observe the deviation of structural dynamics between the S1 and S2 siRNA-Ago2 docked complexes in terms of RMSD and RMSF analysis.
Root mean square deviation represents the measure of deviation from the overlapping of 2 compared structures, and here, each configuration from the MD trajectory file has been compared to the initial configuration structure of the simulation. All the measurements of the RMSD were done based only on the Cα backbone atoms. The RMSD graphs are shown in Figure 5 where RMSD values are plotted against 750 frames (each frame corresponds to 2 pico-second [ps]). Ideally, the RMSD should be zero; therefore, the closer the RMSD is to zero, the more equivalent 2 structures are. It can be seen that compared to S2 docked complex, S1 had lower RMSD values. In case of S1, after 1200 ps, the RMSD values started to go above 2.5 Å, while in case of S2, the RMSD started to go beyond 2.5 Å after only 300 ps.

RMSD of Cα backbone atoms of the Ago2 protein in siRNA-Ago2 complexes: (A) RMSD plot of S1 siRNA-Ago2 complex and (B) RMSD plot of S2 siRNA-Ago2 complex. Ago2 indicates argonaute 2; RMSD, root mean square deviation; siRNA, small interfering RNA.
On the contrary, RMSF represents the portions of a protein structure that are fluctuating from their mean structure by analyzing the average deviation of each protein residue over a period of time from a reference position. The RMSF graphs are shown in Figure 6, and it can be seen that once again S1 docked complex was comparatively more stable than S2 docked complex as in S1 complex the RMSF values only reached 3 Å and in S2 complex, the RMSF values went up to almost 5 Å.
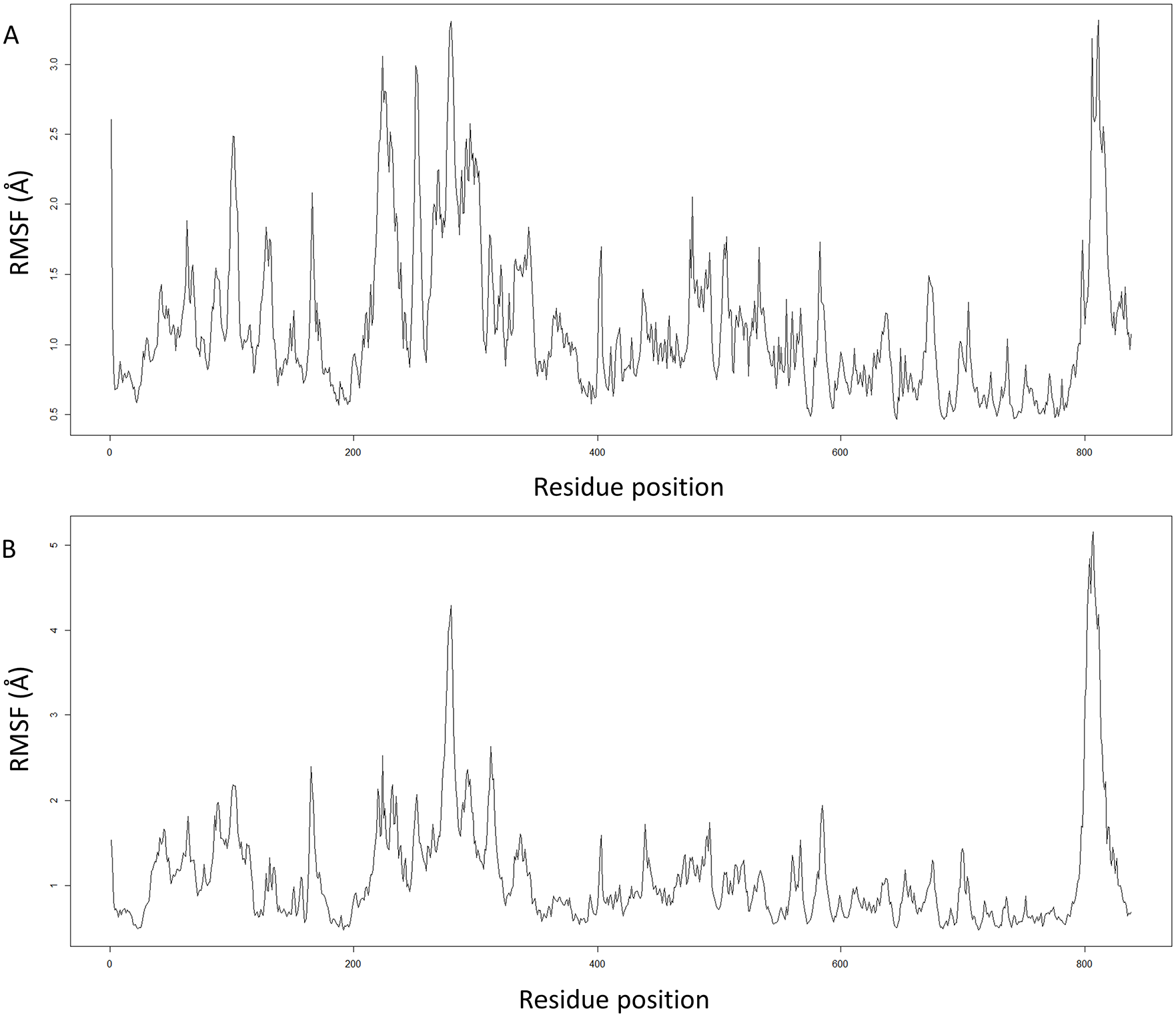
RMSF of each residue of the Ago2 protein in siRNA-Ago2 complexes over 1.5 ns time period: (A) RMSF plot of S1 siRNA-Ago2 complex and (B) RMSF plot of S2 siRNA-Ago2 complex. Ago2 indicates argonaute 2; RMSF, root mean square fluctuation; siRNA, small interfering RNA.
Discussion
RNA interference is a remarkable endogenous regulatory pathway that can bring about sequence-specific gene silencing. If harnessed effectively, RNAi could result in a targeted therapeutic approach with applications ranging from viral diseases to cancer that are particularly hard to address with existing drugs.42,43 The pathways are guided by small RNAs including small interfering RNAs (siRNAs). 44 In addition to translational inhibition and transcript degradation of target mRNAs that are poorly complementary, these effector molecules also guide sequence-specific cleavage of perfectly complementary mRNAs. 44 Theoretically, they can be designed for any gene of interest based on its mRNA sequence alone. Moreover, by using nanoparticles like PEI and other approaches, they can be introduced into the cell for specific knockdown of a gene of interest. Such unlimited potential has particularly made RNAi a favorite gene knockdown strategy in mammalian cells. 45 In this study, we made an attempt through computational methods to rationally design potential siRNA molecules against CLEC5A surface receptor in humans which could lead to new treatment strategies for dengue and JEV infections.
CLEC5A is expressed on the surface of monocytes and macrophages and was first identified by its interaction with DNAX-activating protein of molecular mass 12 kDa (DAP12 or MDL-1). 5 It contains a C-type lectin-like domain in its C-terminal extracellular region and has recently been shown to be a macrophage receptor for DV. The interaction results in the activation of macrophages and a significant release of proinflammatory cytokines, which increases the mortality rate of DV-induced infection. 5 Similar interactions were found between JEV and CLEC5A receptor. After being demonstrated to play a crucial role in the pathogenesis of JEV, CLEC5A is now considered a potential target for new treatments to reduce virus-induced brain damage. 7
In this study, to narrow the search space and improve prediction accuracy, various criteria for predicting and filtering potential siRNA molecules were applied. For instance, siDirect 2.0 predicts off-target minimized siRNA based on an algorithm that takes into consideration the fact that the capability of siRNA to induce off-target effect is highly correlated to the thermodynamic stability of its seed-target duplex. 46 For this, the server first selected highly functional siRNA sequences that follow Ui-Tei, 19 Amarzguioui, 20 and Reynolds 21 rules. For instance they had A/U nucleotides at the 5′ end of the guide strand; G/C nucleotides at the 5′ end of the passenger strand, and at least 4 A/U residues in the 5′ terminal 7 base pair of the guide strand. 19 After that, siRNAs whose seed-target Tm was below 21.5°C for both guide and passenger strands, were regarded almost off-target-free seed sequences. Finally, these siRNAs were checked whether they had at least 2 mismatches to any other nontargeted but near-perfect match transcripts, thus further ensuring that no nontargeted gene silencing can take place. Through these selection steps, siDirect generated and filtrated potential siRNAs.
While these first-generation algorithms heavily depend on base preferences at specific positions, thermodynamic stability, target mRNA secondary structure and several other siRNA features, they can be sometimes not very effective due to their small dataset coverage. However, with the introduction of Huesken’s data set that included 2431 experimentally validated siRNAs, several second-generation algorithms came into the scenario that were based on machine learning models and considered more reliable. Three algorithms were included in this work because they were thought to be the most efficient: s-Biopredsi based on artificial neural network, 23 DSIR 22 and i-score 24 based on linear regression models. Altogether 6 siRNAs were found to give good results in both generations of algorithms and hence predicted to be more efficient during our study.
The GC content of siRNAs is a significant parameter which influences siRNA functionality to a large extent. It has been argued that a high GC content may be a factor that negatively affects the functionality of an siRNA, inhibiting the unwinding of the siRNA duplex, which is necessary for RISC loading. 47 It might also be associated with a prohibitive secondary structure of the target mRNA. On the contrary, several studies have also indicated that a low GC content may lead to decreased functionality, presumably due to reduced target affinity and specificity of the siRNA. 48 For instance, Safari et al 49 showed that the gene silencing efficiency of an siRNA containing 52.6% GC content was higher than another siRNA with 36.8% GC content. As a result, different ranges of GC content have been suggested by different algorithms such as 31.6% to 57.9% 20 and 36% to 52%. 21 In this work, all the candidate siRNAs showed between 33% and 43% of GC content.
Although a low Tm was set to minimize seed-dependent off-target effects, siRNAs designed by different algorithms might still induce off-target effects. These small RNAs can tolerate several mismatches at the mRNA target and thus can silence those targets with imperfect complementarity. Also as they are nearly identical to the related class of miRNAs, they can recognize and degrade mRNAs that are complementary to their seed region and the rest of the nucleotides’ complementarity is insignificant. Therefore, the predicted siRNAs were further checked through BlastN and GESS online tool to ensure their target specificity. BlastN revealed that one of the siRNAs showed 80% query coverage with SMCHD1 gene and hence filtered out from the candidate list. However, because no 3ʹUTR regions were discovered to be over-represented for seed region matches, GESS analysis revealed that none of the off-targeted transcripts were significant using the Fisher exact test and Yates chi-square test. 27 On the contrary, due to the presence of palindromic motifs within the guide strands, 2 other siRNAs were taken off the list. The remaining final 3 candidates showed they were devoid of immunostimulatory motifs 5′-UGUGU-3′ or 5′-GUCCUUCAA-3′ which could stimulate cytokine production, besides having least possibility of off-target effects and less chance of secondary structure formation like hairpin. When sequences contain “GGGG” or “CCCC” motifs they increase the risk of hairpin structures 50 and our predicted siRNAs did not have those motifs.
RNA secondary structure significantly influences the efficiency of a siRNA. Self-structure in either the siRNA sequence or the target mRNA at the binding site may prevent gene silencing 51 by inhibiting RISC-mediated cleavage of target. Therefore, predicting secondary structure of the siRNAs and determining free energy of corresponding folding are crucial for designing effective siRNAs and were conducted in this study. Free-energy minimization approach to RNA structure prediction assumes that the lowest free-energy structure will form. All siRNA molecules were discovered to have positive free energies for folding the guide strands (more than 1.5 kcal/mol), which increased the likelihood that the siRNAs would interact with their targets since they would be less likely to form secondary structures. 52 To further estimate the efficiency of the predicted siRNAs, their free energy of binding with the target mRNA molecules were also measured. Lower binding energy indicates stronger interaction, and therefore well correlates with a better chance of target inhibition. The effectiveness of the siRNAs is also indicated by the higher values of Tm (Cp) and Tm (Conc) as the thermal stability of the RNA molecules is an important parameter of RNAi activity in its conjugated form. 53 Since the Tm values of all the RNAi molecules were above 80°C, it suggested that the molecules were thermally stable and thus efficient. The temperature of the cells may also influence the effectiveness of a particular siRNA since the secondary or local structure of an mRNA can change with temperature changes. One study has found varying effects of the temperature over siRNA efficacy. 54 While some siRNAs showed no significant changes or slightly increased silencing at lower temperatures, others showed significantly reduced or no silencing when the cells were grown at lower temperatures like 33°C and 35°C. CLEC5A receptor is expressed on tissue macrophages and they are often exposed to elevated tissue temperature during inflammation and fever. Since an increased temperature has not been reported to significantly affect gene silencing, it is assumed that the predicted siRNAs from our study would be able to retain their silencing efficiency in the human body.
The downregulation potential of a candidate siRNA crucially depends on its accessibility of the target site within the mRNA molecule. 55 Hence, this benchmark was also studied in detail in this work using the Sirna module of S-fold. Target site accessibility score ranges between 0 and 8, where 0 indicates antisense siRNA binding energy is greater than −2 kcal/mol and 8 indicates the binding energy is less than −16 kcal/mol. The high target site accessibility score for every siRNA in this study, therefore, indicated stronger target binding (Supplementary Table 1).
The key component of RNAi machinery is the Ago2 protein which plays an important role in the functioning of siRNAs. The interactions between the candidate siRNA molecules and Ago2 protein were analyzed in this study. Human Ago2 protein consists of 4 domains including N-terminal (Asp53-Ser139), PAZ (Pro229-Val347), MID (Gly445-Pro580), and catalytic PIWI (Gln581-Ala859). 56 The PAZ domain holds the 3′-end of siRNA while the MID domain binds to the 5′-end. It was found that in each of the docked complexes, ARG714, ARG792, ARG68, SER220, TYR529, CYS546, HIS600, ALA603, SER798, GLN757, LYS566, THR559 residues among others formed hydrogen bonds with the siRNAs. On the contrary, ARG68, ARG97, ARG375, LYS709, ARG761, ARG792, and HIS600 formed electrostatic interactions while LYS525 and CYS66 residues formed hydrophobic bonds with all the siRNAs. Some of these residues including TYR529, Arg761, SER798, ARG792, LYS566, and CYS546 have also been previously reported to interact with other siRNAs and miRNAs.56,57
S1 showed the strongest interaction with the Ago2 protein as revealed by the lowest docking energy score as well as a large number of hydrogen bonds. The score reflects the potential energy change when the protein and ligand come together. A positive scoring function represents higher potential affinity. Since in molecular docking analysis of a complex, the energy of the complex is subtracted from its components, a low and negative score indicates that the complex is more stable than its components by themselves. Therefore, they would have a favorable binding interaction. This means that a very negative score corresponds to a strong binding and a less negative or even positive score corresponds to a weak or nonexisting binding. When the interaction between S1 siRNA and Ago2 protein was explored, it was found that with the PAZ domain C20 formed 1 and U21 formed 3 hydrogen bonds; G16 formed 1 electrostatic bond and U21 formed 2 hydrophobic bonds. On the contrary, with the MID domain, U1 formed 4 hydrogen bonds, U2 formed 2 hydrogen bonds and U3 formed 1 hydrogen bond, U1 also formed 2 electrostatic and 3 hydrophobic bonds. To further examine the stability of the protein-siRNA complexes, MD simulations were performed on S1 and S2 docked complexes. Performing MD simulations over real experiments allow us to obtain information at the microscopic level and later express it in macroscopic properties. 58 It was revealed from the RMSD and RMSF analyses that compared to S2, S1 siRNA formed a more stable complex with Ago2 protein which also well correlated with its binding energy and hydrogen bond measurements.
All in all, in this study with the help of multiple computational tools, the most effective and potential siRNA molecules have been designed against the CLEC5A receptor in humans. In the next step, the efficacy of these predicted siRNAs needs to be experimentally validated. The candidate siRNAs may prove instrumental in the downregulation of the viral activity in tissue macrophages through transient silencing of the receptor and therefore may lead to a promising new therapeutic approach against dengue and JEV.
Conclusion
RNA interference molecules were designed through in silico methods against the CLEC5A receptor of humans to silence their expression. We selected 3 siRNAs as final candidates because they showed good target accessibility, positive energy of folding, and strong binding with Argonaute2 protein. The stability of the siRNA-Ago2 docked complexes was also analyzed through MD simulation. The approach may pioneer a vision for the chemical synthesis of novel antiviral therapeutics for the treatment of dengue and JEV at the genomic level. Currently, ONPATTRO and GIVLAARI RNAi therapeutics are commercially approved on the market for the treatment of polyneuropathy and acute hepatic porphyria, respectively. We hope this study will help to develop a similar treatment strategy for dengue and JEV infections.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322221142122 – Supplemental material for In Silico Design of siRNAs for Silencing CLEC5A Receptor as a Potential Therapeutic Approach Against Dengue and Japanese Encephalitis Virus Infection in Human
Supplemental material, sj-docx-1-bbi-10.1177_11779322221142122 for In Silico Design of siRNAs for Silencing CLEC5A Receptor as a Potential Therapeutic Approach Against Dengue and Japanese Encephalitis Virus Infection in Human by Tahirah Yasmin, Maisha Adiba, Abdullah Al Saba and AHM Nurun Nabi in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
TY and AHMNN conceived the project. TY collected the data. TY, MA and AAS performed the analyses. TY and AHMNN wrote the manuscript. The manuscript was reviewed by all authors.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.