
Abstract
MicroRNAs (miRNAs) are small, noncoding RNA molecules that regulate transcriptional and posttranscriptional gene regulation of the cell. Experimental evidence shows that miRNAs have a direct role in different cellular processes, such as immune function, apoptosis, and tumorigenesis. In a viral infection context, miRNAs have been connected with the interplay between host and pathogen, occupying a major role in pathogenesis. While numerous viral miRNAs from DNA viruses have been identified, characterization of functional RNA virus-encoded miRNAs and their potential targets is still ongoing. Here, we used an in silico approach to analyze dengue Virus genome sequences. Pre-miRNAs were extracted through VMir software, and the identification of putative pre-miRNAs and mature miRNAs was accessed using Support Vector Machine web tools. The targets were scanned using miRanda software and functionally annotated using ClueGo. Via computational tools, eight putative miRNAs were found to hybridize with numerous targets of morphogenesis, differentiation, migration, and growth pathways that may play a major role in the interaction of the virus and its host. Future approaches will focus on experimental validation of their presence and target messenger RNA genes to further elucidate their biological functions in human and mosquito cells.
Keywords
Introduction
Dengue virus (DENV) is an enveloped, mosquito-borne virus with a single-stranded RNA genome of positive polarity and is a member of the Flaviviridae family, specifically belonging to the flavivirus genus. This genus contains nearly 80 viruses distributed worldwide and includes important human pathogens such as yellow fever virus, Japanese encephalitis virus, West Nile virus (WNV), and tick-borne encephalitis virus (TBEV).1,2 DENV is classified into four distinct antigenic types (DENV −1 to −4). In tropical and subtropical environments, DENV infection is an issue of critical importance for public health. Every year, millions of people are infected, producing mild to debilitating febrile illness or a more aggressive disease that can cause hemorrhagic episodes, vascular leakage, severe thrombocytopenia, and shock. Nevertheless, the molecular mechanisms involved in the pathogenesis of DENV remain poorly understood and there is no effective vaccine/drug available to reduce its symptoms or inhibit the infection based on any of the four serotypes. In recent years, due to anthropogenic intervention in natural habitats, the geographical range of DENV has been extended and more cases have been reported.3,4
DENV enters its target cells (primarily dendritic cells, monocytes, and macrophages) via receptor-mediated endocytosis in a clathrin-dependent manner. The viral genome consists of a single, long open reading frame (ORF) flanked by 5′ (100 nt) and 3′ (∼400 nt) untranslated regions (UTRs). 5 8 The genome has a positive-sense polarity and is translated at the rough endoplasmic reticulum, giving rise to a polyprotein. It is cotranslationally and posttranslationally cleaved into three structural proteins (C, prM, and E), constituting the building blocks of the virion, and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) that are involved in RNA replication, assembly, and modulation of the immune response of the host cell.5,6,8,9
MicroRNAs (miRNAs) are small RNA molecules that are known for their important role as gene regulators in metazoan. In general, miRNAs are processed from structured RNAs into single-stranded molecules, 18-23 nt in length, which have been shown to control translation of messenger RNAs by cleaving or blocking the target. In the mammalian cytoplasmic context, miRNAs attach to their target with incomplete complementarity in association with cellular proteins commonly called the RNA-Induced Silencing Complex (RISC). Regulation mediated by miRNAs is important in an extensive range of biological processes, including growth, development, cellular division, and oncogenesis. 10 12
In a viral infection panorama, miRNAs have been associated with the interplay between host and pathogen, playing a major role in viral pathogenesis. It is clear that studies on host-pathogen interactions at the miRNA level are still in their infancy and seem to be a promising field for the study of the pathogenicity of human viruses. Viral miRNAs can regulate both cellular and viral gene expressions through the regulation of cellular elements involved antiviral responses, simulating cellular miRNAs, or targeting their own viral messenger RNAs to modulate the viral replication cycle. 13 15
Remarkably, most current knowledge about viral miRNAs is derived from DNA viruses, primarily from the Herpesviridae family. For cytoplasmic RNA viruses, miRNAs were hypothesized to be absent because of the potential degradation of the viral RNA genome during the excision of virus-encoded miRNA precursors (pre-miRNAs). 14 However, it has been revealed that functional, virus-derived miRNAs can be processed by cytoplasmic RNA viruses when a pre-miRNA sequence is introduced into the virus genome. Good examples are the Sindbis virus (SINV) and TBEV. 16 18 A pri-miR-124 was incorporated into the viral genome of the SINV and was able to produce pre-miR-124 and miR-124 in a DGCR8-independent, exportin-5-independent, and Dicer-dependent manner. 17 Additionally, the flavivirus TBEV was shown to express an Epstein-Barr virus miRNA through the integration of its miR-BART2 precursor in the 3′-UTR of the TBEV genome.16,18
A special case is the WNV. This virus has a highly conserved stem-loop (SL) region in the viral 3′-UTR that serves as a source for the generation of a mature miRNA in infected mosquito cells. This miRNA, called KUN-miR-1, targets mosquito GATA4 mRNA that leads to its up-regulation in cells. Inhibition of KUN-miR-1 or depletion of GATA4 mRNA both led to reduced WNV RNA replication.13,19
Identification of animal and plant miRNAs is mostly based on homology methods; however, the same strategy cannot be followed with RNA viruses. The high mutation rates and the lack of validated miRNAs impede an adequate and accurate prediction. To increase the amount of information related to viral miRNA discovery in RNA virus, we methodically scanned pre-miRNAs derived from DENV genomes. Then, we filtered the results, validated the secondary structure and identified mature miRNAs. We also predicted the potential targets of those miRNAs and their structures and functions. Overall, we carefully produced a working methodology for identifying miRNAs and their potential cellular targets, which can be used with other flaviviruses or RNA viruses. The workflow of our investigation is shown in Figure 1.

A synopsis of the used methodology during miRNA prediction of DENV2.
Methods
Viral Sequences and the Prediction of Pre-miRNAs by an ab Initio Approach
For this work, we used two different genome sequences of DENV serotype 2 downloaded from publicly available databases, which included the ORFs and the 5′ and 3′-UTRs of the viral RNA [GenBanK accession numbers: JF357906.1 and FJ390389.1]. VMir was used to analyze the DENV2 sequences for possible pre-miRNA hairpin structures, using stringent filtering parameters as described in other publications.
13
The VMir program (downloadable online) uses several predefined but adjustable parameters. This
Identification of Putative Pre-miRNA Sequences and Mature miRNAs
To discriminate real pre-miRNAs from other hairpin structures with similar SLs (SL pseudo hairpins), we employed the ncRNA Feature Extraction and pre-miRNA Classification Web Tool (accessible at http://150.140.142.24:82/PredictionAnon.aspx), which decides whether each hairpin is either a pseudo-pre-miRNA-like hairpin or a real pre-miRNA using a Support Vector Machine classifier (SVM). 21
With the purpose of extracting mature miRNA:miRNA* duplexes from pre-miRNA hairpins, we used the MaturePred Web Tool (accessible at http://nclab.hit.edu.cn/maturepred/). This software also uses a model based on SVM that predicts the starting position of an miRNA by performing discriminant analysis against the query hairpin structure using various features of known real/pseudo miRNA:miRNA* duplexes as a training set (position-specific features, energy-related features, structure-related features, and stability-related features). 22
Secondary Structure Validation
Pre-miRNA sequences were submitted to Mfold (accessible at http://mfold.rna.albany.edu) to check the fold-back secondary structure. The default parameters for Mfold were used, and all qualifiers were recorded, including the nucleotide length, position of the matching regions, the number of arms per structure, and the Minimal Folding Free Energy (MFE, AG kcal/mol). We also calculated the Minimal Folding Free Energy Index (MFEI) as previously described.23,24
Prediction of Potential Targets and Functional Enrichment Analysis
Human UTRs were downloaded from the UTRdb database. Subsequently, the potential 3′-UTR targets for the putative miRNAs were scanned with the assistance of the Linux-based miRanda software. 25 This software operates thermodynamics and dynamic programming alignments, along with statistical parameters, for target prediction against the human genome. The parameters assigned for miRanda hybridization included a default alignment score ≥ 140 and an MFE for an miRNA:miRNA* duplex of ≤–35 kcal/mol, and the other parameters were kept as default. 26 The matched UTRs were submitted to the NCBI BLAST platform to visualize the genome context, and the biological function was annotated. The diverse steps involved in the identification and target prediction of the miRNAs from DENV are presented in Figure 1. In order to enrich the identified genes with connection to specific functional terms, the potential targets were analyzed using Cytoscape software and its plug-in: ClueGo, applying the Gene Ontology database (released January 2014). Ontologies were designated as biological processes, immune system processes, reactome, molecular function, and cellular component. Enrichment was executed by the right-sided hyper-geometric test and its probability value was corrected by the Bonferroni's step-down method. For every procedure, the maximum stringency filters were utilized to minimize the noise and background results, thereby guaranteeing the fewest number of false-positive results.
Results and Discussion
Prediction of Pre-miRNA in DENV
Many mature miRNAs are evolutionarily conserved from organism to organism, which simplifies the prediction of the existence of new miRNAs in other species.27,28 However, other approaches should be evaluated for fast-evolving biological entities such as viruses. Using an

Predicted pre-miRNA secondary structures. Mature miRNA sequences are presented in red.
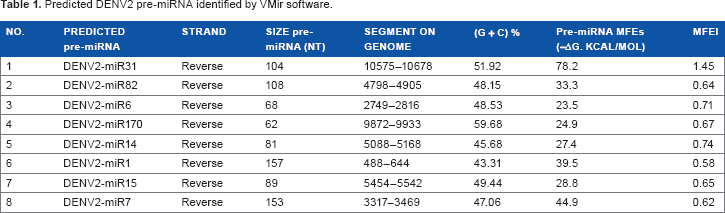
Predicted DENV2 pre-miRNA identified by VMir software.
Mature miRNAs
Comparable to other biological systems, DENV miRNAs can be located on either of the two arms in the secondary hairpin structure (Fig. 2). Of the eight miRNAs identified in DENV2, three are located in the 5′-arm of the SL hairpin structure, while five are in the 3′-arm (Table 2). The existence of this feature is also present in animal miRNAs, where mature miRNA can be processed from either of the two arms of the SL hairpin secondary structure. 23 The length of the mature miRNAs was the same because it was a controlled and recommended parameter for the prediction of sequences in non-plant organisms. Remarkably, just three mature miRNAs start with a 5′-terminal uridine residue, a typical characteristic of miRNAs recognized by the AGO1 protein. These findings indicate that the predicted miRNAs may be noncanonical. 31
Characteristics of DENV2 mature miRNAs.

Prediction of the Potential Targets for Putative miRNAs in DENV and Functional Annotation
A better understanding of the dynamics between miRNAs and their targets will help to understand the complexity of biological regulation and other aspects of virus-host dynamics. In silico prediction of miRNA targets provides a good alternative for identifying potential target sites based on their complete or partial complementarity with the miRNAs. Nevertheless, it is well known that in animal systems, miRNA targets are difficult to predict because miRNA and mRNA pairs frequently contain several mismatches, gaps, and G + U base pairs in many positions. 26
In this study, pairwise comparison of whole human 3′-UTRs against eight mature miRNA of DENV has been conducted. The miRanda algorithm was used, which incorporates thermodynamic stability calculations of miRNA:mRNA* duplexes and alignment procedures for detecting the probable binding site on the 3′-UTRs. We observed 53 transcript targets for eight DENV miRNAs (Table 3). The predicted targets for a single miRNA vary from just one (DENV2-miR31, DENV2-miR6, DENV2-miR170, and DENV2-miR1) to >10 (DENV2-miR14, DENV2-miR15). While several miRNAs may regulate just one mRNA, these results demonstrate another type of regulation where a single miRNA can regulate numerous transcripts, elevating the complexity of cellular processes.
Predicted miRNAs targets indetified by

Gene ontology has become a very useful tool for the mining of gene/protein datasets and their functional annotations. The functional enrichment analysis was conducted using the ClueGo plug-in on the potential targets of the DENV miRNAs. The 53 genes were significantly enriched to two main functional clusters, ie, anatomical structure formation involved in morphogenesis (10 terms) and cell projection morphogenesis (13 terms) (Fig. 3). Many of the terms obtained from the annotation were related to development, movement, differentiation and migration. Clearly, experimental confirmations are needed to validate these targets under infection conditions.

Functional enrichment analysis of the predicted targets of the Dengue Virus miRNAs.
Conclusions
The importance of miRNAs in cellular systems is clear. However, understanding of the role for viral miRNAs is just in its infancy. Viral miRNAs can be extraordinary tools to modulate cellular or viral gene expression. Their nonimmunogenic nature, small size, high specificity, and capacity for multiple transcript regulation are reasons to expect their presence in RNA viruses. In addition, the picture becomes more complex when the search is extended to include viral replication intermediates. In this case, the presence of viral miRNAs in the negative-sense RNA used as a replication template for the viral genome may be a new source of information, which could explain pathogenesis-related phenomena.
The outcomes of the in silico predictions are useful to guide experimental design to achieve biological validation. The next logical step after these types of analyses is to scrutinize whether miRNAs can be processed in eukaryotic cells (mammalian and mosquito cell lines). In summary, we performed a complete scan of the DENV genome and its replication intermediates to obtain all putative pre-miRNAs for the entire sequence that were later filtered and matured by machine learning web tools. A total of eight miRNAs were identified, and these miRNAs share comparable features with other known miRNAs (animal miRNAs); however, other characteristics, such as the absence of a uridine at the 5′ end, reveal possible noncanonical processing and different biogenesis. In silico target prediction generated 53 genes. The potential targets are involved in different important biological processes as anatomical structure formation involved in morphogenesis and cell projection morphogenesis, suggesting that they could play an important role in the process of virus-host interactions during infection.
Author Contributions
MO carried out the experimental procedures and data analysis. MO, NC, JPF, and JCG contributed with the writing of the manuscript. JCG designed the project from which this manuscript was planned. MO, NC, JPF, and JCG agreed with manuscript results and conclusions. All authors read and approved the final manuscript.
Supplementary Data
Supplementary File 1
VMir analysis of the Dengue Virus genome (A) isolate DENV-2/NI/BID-V3227/2008 and (B) New Guinea C derivative strain; shown are all hairpins that fold in a 500 nt window and achieved a VMir score of 115 or above.