
Abstract
Reutealis trisperma oil is a new source for biodiesel production. The predominant fatty acids in this plant are stearic acid (9%), palmitic acid (10%), oleic acid (12%), linoleic acid (19%), and α-eleostearic acid (51%). The presence of polyunsaturated fatty acids (PUFAs), linoleic acid, and α-eleostearic acid decreases the oxidation stability of R. trisperma biodiesel. Although several studies have suggested that the fatty acid desaturase 2 (FAD2) enzyme is involved in the regulation of fatty acid desaturation, little is known about the genetic information of FAD2 in R. trisperma. The objectives of this study were to isolate, characterize, and determine the relationship between the R. trisperma FAD2 fragment and other Euphorbiaceae plants. cDNA fragments were isolated using reverse transcription polymerase chain reaction (PCR). The DNA sequence obtained by sequencing was used for further analysis. In silico analysis identified the fragment identity, subcellular localization, and phylogenetic construction of the R. trisperma FAD2 cDNA fragment and Euphorbiaceae. The results showed that a 923-bp partial sequence of R. trisperma FAD2 was successfully isolated. Based on in silico analysis, FAD2 was predicted to encode 260 amino acids, had a domain similarity with Omega-6 fatty acid desaturase, and was located in the endoplasmic reticulum membrane. The R. trisperma FAD2 fragment was more closely related to Vernicia fordii (HM755946.1).
Introduction
Reutealis trisperma (locally named as Kemiri Sunan) is a member of the family Euphorbiaceae that originates from the Philippines and has been cultivated in many tropical countries, including Indonesia, Malaysia, and India.1,2 This plant exhibits good adaptation to drought and can grow on marginal land or ex-mining land. 3 R. trisperma is currently being developed by the Indonesian government as a raw material for biodiesel production. The productivity of R. trisperma reaches 6 to 8 tons of biodiesel per hectare per year. 3 However, R. trisperma has seeds that are poisonous because of their 51% non-edible fatty acids. This toxicity is caused by the presence of α-eleostearic acid.4,5 Cellular lipids are mainly composed of fatty acids that act as components of the membrane, signaling molecules in plant response mechanisms to stress and defense, and energy sources.6-8 Lipids in plants are stored as triacylglycerols, which are the main components of vegetable oils. 9 Vegetable oil is widely used in the fields of food, industry, and biodiesel production.
Biodiesel results from the transesterification of triacylglycerol with methanol.10-12 Lipids composed of fatty acids are distinguished by the presence or absence of double bonds, namely, saturated and unsaturated fatty acids. Unsaturated fatty acids are further categorized based on the number of double bonds in the carbon chain, namely, monounsaturated fatty acids (MUFAs) and polyunsaturated fatty acids (PUFAs). 13 The composition of fatty acids in vegetable oils is an important factor in determining the quality of biodiesel. Increasing the number of double bonds in the chemical structure of PUFAs causes biodiesel to be susceptible to oxidation, affecting the performance of diesel engines. Good quality biodiesel contains a higher number of MUFAs than saturated fatty acids and PUFAs. 14 Recently, non-edible R. trisperma oil has been considered as a new carbon source for biodiesel production. 15 The dominant fatty acids in R. trisperma are stearic acid (9%), palmitic acid (10%), oleic acid (12%), linoleic acid (19%), and α-eleostearic acid (51%). 4 Linoleic acid is predominantly found in plant oils.16-18 High PUFA levels cause the oxidation stability test of R. trisperma biodiesel to fail. A previous study showed that the oxidation stability of R. trisperma is only 1.5 hours from the standard 6-hour test. 1 Increasing the number of double bonds in vegetable oil (PUFA) is inversely proportional to the quality of biodiesel; therefore, research on the characterization of key enzymes in lipid metabolism is critical.
To date, 2 key enzymes have been identified and characterized: diacylglycerolacyl-CoA acyltransferases (DGATs) and fatty acid desaturase (FAD). The DGAT enzyme is an important enzyme that limits the rate of accumulation of plant lipid storage, while the FAD enzyme functions in the introduction of double bonds in the hydrocarbon chain of the fatty acid chain.18,19 Polyunsaturated fatty acid biosynthesis is catalyzed by the fatty acid desaturase 2 (FAD2) enzyme, which forms double bonds in oleic acid (C Δ12 atom) to become linoleic acid. 20 The modification of enzyme activity is necessary for improving the value of a plant, especially for improving the quality of biodiesel. Genetic engineering by silencing the FAD2 gene to increase the oleic acid content and simultaneously reduce linoleic acid levels has been carried out on Brassica napus, Gossypium hirsutum, and Arachis hypogaea.21-23
The modification of enzyme expression and activity is related to the genetic information of the genes encoding the enzyme. The FAD2 gene has been identified in several plant species, including Vernicia fordii, Jatropha curcas, Ricinus communis, A. hypogaea, Arabidopsis thaliana, Helianthus annuus, B. napus, and Cucurbita pepo. 19 However, genetic information for FAD2 R. trisperma is currently unknown; therefore, it is necessary to research the FAD2 gene in R. trisperma as a basis for exploring gene function and applying genetic engineering to improve oil composition in R. trisperma. In this study, we isolated and characterized FAD2 fragments, which play a role in the biosynthesis of unsaturated fatty acids in R. trisperma.
Materials and Methods
Plant materials
R. trisperma plants and seeds were obtained from the Purwodadi Botanical Garden of the Indonesian Institute of Science. Young leaves and seeds of the plant were stored at –20°C before being used for further experiments.
Data collection of FAD2
Fatty acid desaturase 2 nucleotide sequences were obtained from GenBank. The nucleotide sequences originate from the Euphorbiaceae family, which produces FAD2. Several Euphorbiaceae species, H. brasiliensis (accession number: DQ023609.1), R. communis (accession number: NM_001323719.1), J. curcas (accession number: NM_001308778.1), and V. fordii (accession number: AF525534.1) were selected. DNA sequence alignment was conducted using the CLC Sequence Viewer 7 program (QIAGEN Digital Insights, https://digitalinsights.qiagen.com). Degenerate primers were designed based on the conserved region obtained from DNA sequence alignment analysis. Primer analysis was performed using OligoAnalyzer 3.1 (http://sg.idtdna.com/calc/analyzer).
Total RNA extraction and cDNA synthesis
Total RNA extraction was carried out using the Plant Total RNA Mini Kit (Genaid) according to Jadid et al. 24 The seeds were peeled, and the endosperm was thinly dissected and immediately placed into a 1.5-mL tube floating on liquid nitrogen. Total RNA concentration was quantified using a Nanodrop spectrophotometer (ND-1000, Thermo Scientific, USA). Total RNA (0.21 µg) was used for first-strand cDNA synthesis using an AffinityScript qPCR cDNA Synthesis Kit (Agilent Technologies, USA), following the kit instructions with an oligo (dT)20 primer.
Isolation of FAD2 cDNA fragments
Reverse transcription (RT) polymerase chain reaction (PCR) amplification was conducted using RT-PCR (Platinum Green Hot Start PCR 2× Master Mix, InvitrogenTM, Thermo Fisher Scientific, USA) and a pair of sense and antisense degenerate oligonucleotides, designed based on the conserved nucleotide region of known FAD2 sequences. Polymerase chain reaction was performed using an initial denaturation step at 94°C for 2 minutes, followed by 35 cycles at 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute, and a final extension at 72°C for 5 minutes. Several FAD2 primers were designated as follows: F10 forward primer (5′-ATGGGWGCYGGTGGYAGAATGTCWG-3′), F15 reverse primer (5′-KATARTGYGGCATTGTWGARAAC-3′), and F12 forward primer (5′-CCTTAYTTTTCATGGAAACCAYAGC-3′). The amplicons obtained from the previous PCR were then migrated using 2% agarose gel electrophoresis and subsequently purified using Promega Wizard® SV Gel and PCR Clean-Up System (DNA Purification by Centrifugation). Finally, the purified DNA fragments were subjected to sequencing analysis.
In silico analysis of FAD2 fragments
The nucleotide sequence was opened using the BioEdit software. The sequenced data file was converted into a FASTA data file and then saved to the CLC Sequence Viewer 7 program (QIAGEN Bioinformatics). The edited nucleotide sequences were then analyzed using Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/), open reading frame (ORF) prediction using the NCBI ORF Finder online program, and a conservative domain search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). These predictions were used to determine the identity of the isolated DNA fragments and the level of similarity of the FAD2 with other plants.
The FAD2 sequence of R. trisperma was aligned with the sequence of FAD2 from the other selected species. The alignment was performed using the ClustalW alignment algorithm (MEGA 7.0). Phylogenetic tree constructions were prepared using software similar to the neighbor-joining algorithm with a bootstrapping value of 1000×. Prediction of subcellular location was performed by detecting the signal peptide of an amino acid sequence using the TargetP 1.1 program (www.cbs.dtu.dk/services/TargetP). TargetP results provide 4 location predictions in the “Loc” column, namely, chloroplasts (C), mitochondria (M), secretory pathway (SP), or other locations (“_”). If the TargetP value indicates another location prediction (“_”), it is necessary to check it using the TMHMM server program (version 2.0; http://www.cbs.dtu.dk/services/TMHMM/) to determine whether the associated protein is a membrane protein. 25
Data analysis
The obtained data were analyzed descriptively. Data included DNA sequences that were analyzed for BLAST homology, ORF predictions, conservative domains against the GenBank database, signal peptide prediction data, transmembrane domains, and phylogenetic tree construction.
Results and Discussion
Isolation of FAD2 fragments from R. trisperma
The isolation of the FAD2 R. trisperma gene was carried out using the RT-PCR approach. 26 The RT-PCR method is a genomic DNA amplification technique that is preceded by an enzymatic change of mRNA to cDNA. 27 The first strand of cDNA was synthesized using the reverse-transcriptase enzyme and random hexamer primers (oligo dT), 28 allowing cDNA synthesis from polyadenylated RNA simultaneously. 29 The results of RNA extraction on R. trisperma seeds yielded a total RNA concentration of 53.4 ng/µL. Furthermore, the synthesis of the first strand of cDNA was carried out using 0.21 µg of total RNA. The amplification of FAD2 fragments was performed using FAD2-specific primers. In this study, degenerate primers were used because of the unavailability of genetic information data for R. trisperma FAD2 in GenBank. 30 Degenerate primers were designed by gathering genetic information for FAD2 from several other plants. FAD2 sequence alignment was carried out on 4 Euphorbiaceae species, namely, J. curcas, V. fordii, R. communis, and H. brasiliensis, to determine the conservative region.31,32 The conserved region that showed the lowest degeneracy was used as the basis for primary selection because it has a higher specificity. 33
In the present study, we successfully amplified 2 FAD2 fragments using the FA10-FA15 and FA12-FA15 pair of primers (Figure 1). The longest fragment (983 bp) was obtained from DNA amplification using the FA10-FA15 primer combination. Another fragment was 585 bp in length. The 983-bp fragment was sequenced. The Sanger sequencing chromatogram showed that the first 10 to 30 nucleotides are of low quality and can be ignored because it is the effect of the chemical sequencing reaction. 34 By comparing the FAD2 sequence from other plant species, we found that our sequence still needs to be completed. Therefore, complete cDNA isolation of FAD2 using the rapid amplification of cDNA ends (RACE) method might be performed in the future.35-37
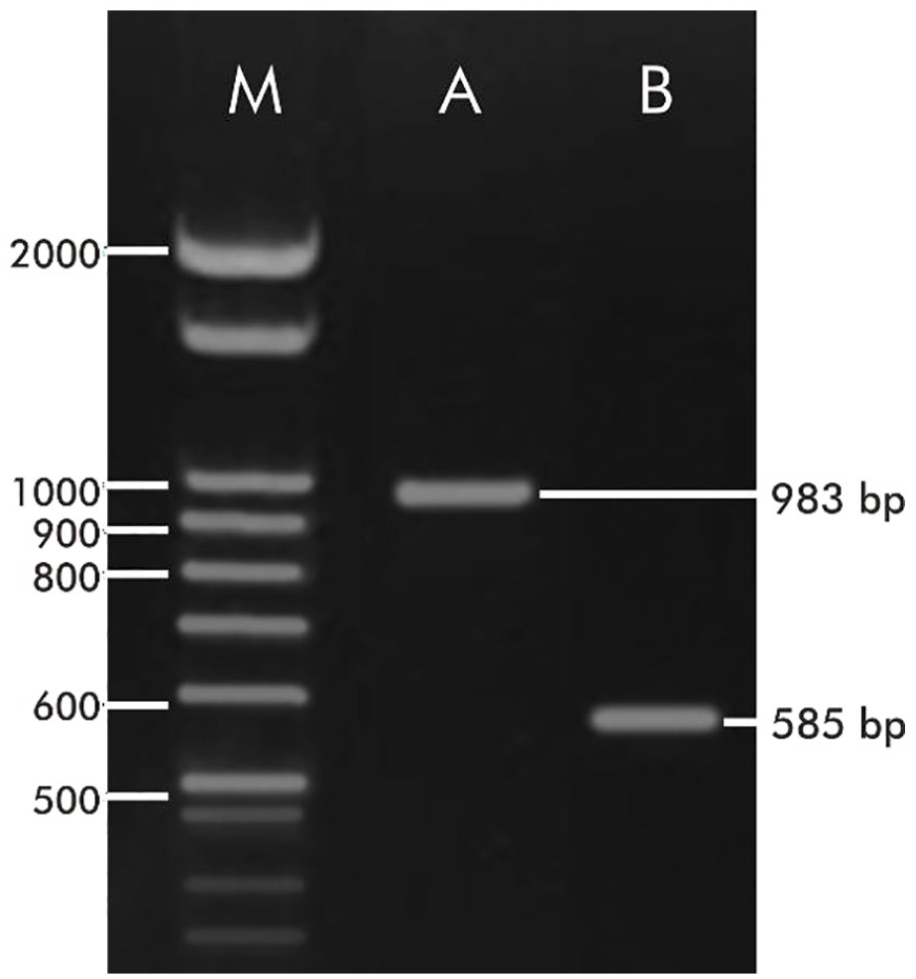
Electropherogram of FAD2 from Reutealis trisperma. M: Marker; A: fragment obtained with FA10-FA15 pair of primer; B: fragment obtained with FA12-FA15 pair of primer.
Homology sequence and conserved domain analysis of the R. trisperma FAD2
The nucleotide analysis of R. trisperma using BLAST (Table 1) showed an E-value of 0 against several Euphorbiaceae species, demonstrating the high degree of similarity between R. trisperma sequences and these species. 38 The sequence of R. trisperma FAD2 can cover 99% of nucleotides with a similarity level of 86% to 96% to several other Euphorbiaceae species. It can be assumed that several R. trisperma nucleotides are not the same as the Euphorbiaceae species deposited in GenBank. 39 The fragment of R. trisperma FAD2 showed the highest percentage of similarity to V. fordii FAD2 (accession number HM755946), which was 96%, and the lowest percentage of similarity to Manihot esculenta FAD2 (accession number XM_021766109.1), which was 86%, indicating that the R. trisperma FAD2 sequence is highly similar to that of V. fordii FAD2. The UniProt database also demonstrated that the similarity level of FAD2 in the Euphorbiaceae family was 90%. Based on this, it can be concluded that the 923-bp gene fragment that was isolated is thought to be the FAD2 gene, but this needs to be confirmed by conservative domain analysis.
Similarity sequence analysis of FAD2 from Reutealis trisperma with other Euphorbiaceae species.

Abbreviation: FAD2, fatty acid desaturase 2; nt|aa, nucleotide|amino acids.
FAD2 is an enzyme that plays a vital role in the formation of double bonds between oleic acid and linoleic acid. 35 Enzymes are proteins or amino acid polymers with high specificity for other molecules and function as reaction catalysts that can form and break covalent bonds. 40 The activity of a protein is regulated by a domain that usually has a specific function. 41 Domain analysis was carried out through the Conserved Domain Database program at the National Center for Biotechnology Information (NCBI), using amino acid sequences as initial data. Therefore, the amino acid composition of the R. trisperma FAD2 fragment must be determined first. Prediction of amino acid composition by in silico analysis can be performed using the NCBI ORFfinder program. Based on the ORFfinder program, a nucleotide sequence of 783 bp was obtained, which encoded 260 amino acids (Table 2). The amino acid sequences obtained were then reused for conservative domain prediction using the conserved domain database program.
Open reading frame prediction of the FAD2 fragment from Reutealis trisperma.

Abbreviation: FAD2, fatty acid desaturase 2.
Conservative domain analysis showed that FAD2 of R. trisperma possesses a high similarity to omega-6 FAD with an E-value of 0 (Table 3) and a Δ12 fatty acid desaturases (Δ12-FADs)-like domain (Figure 2). The results of this in silico analysis are in accordance with Dong et al, 42 which states that the FAD2 enzyme is included in omega-6 FAD; therefore, it can be concluded that the successfully amplified DNA fragment is the R. trisperma FAD2 gene fragment.
List of the conserved domain of the Reutealis trisperma FAD2.

Abbreviation: FAD2, fatty acid desaturase 2.

Conserved domain of the Reutealis trisperma FAD2 fragment. FAD2 indicates fatty acid desaturase 2.
The conserved domain of Δ12-FADS-like includes integral-membrane enzymes, namely, Δ12 acyl-lipid desaturases, oleate 12-hydrolases, omega-3, and omega-6 FADs. This enzyme is found in various organisms, including high-level oil-producing plants, 35 algae, 43 diatoms, 44 yeast, 45 and bacteria. 46 The enzyme activity is influenced by temperature. Menard et al 47 reported that a decrease in temperature could increase the levels of fatty acid desaturation in membrane lipids, subsequently changing membrane fluidity. According to Zauner et al, 48 the characteristic domain of the membrane-integral desaturase enzyme is the presence of 3 histidine residues (HXXXH, HXX (X) HH, and HXXHH). Residual histidine acts as a site for oxygen and substrate oxidation. The enzyme FAD2 (Δ12-FADS; omega-6 FAD) is a multifunctional membrane-integral enzyme that catalyzes the formation of trans- or cis-double bonds at position Δ12 in oleic acid. 19 In addition, it is also stated that the motive for histidine residues in FAD2 is HECGHH, HRRHH, and HV [A/C/T] HH. 19 Meanwhile, the histidine residue motifs in R. trisperma FAD2 that were found through the conserved domain program were HECGHH and HRRHH. The HV [A/C/T] HH motif was not found in the amino acid sequence of R. trisperma FAD2.
Residual histidine has an essential catalytic function and acts as a ligand for 2 Fe atoms. 16 Mutations in one of the histidine residues can weaken ionic bonds and reduce the catalytic activity of the enzyme. 19 The Fe atom, which binds to the histidine residue, plays a vital role in the catalytic mechanism of fatty acid desaturation (Figure 3). The diiron centers are in an oxidized state (diferric or FeIII-FeIII) and connected to the µ-oxo bridge at rest. The reduction of both Fe ions by transferring electrons of the 2 ferredoxins gives the reduced form of Fe (diferrous or FeII-FeII). The reduced enzymes bind molecular oxygen, resulting in the formation of the peroxo intermediate. Cleavage of the O–O bond results in the formation of an active central diiron “Q” (diferryl or FeIV-FeIV). The active central diiron performs hydrogen abstraction and requires energy from the inactive fatty acid methylene groups to produce radical intermediates. The loss of the second hydrogen results in the formation of a double bond and is accompanied by the release of H2O and regeneration of the active site, which is oxidized, and the µ-oxo bridge. 16
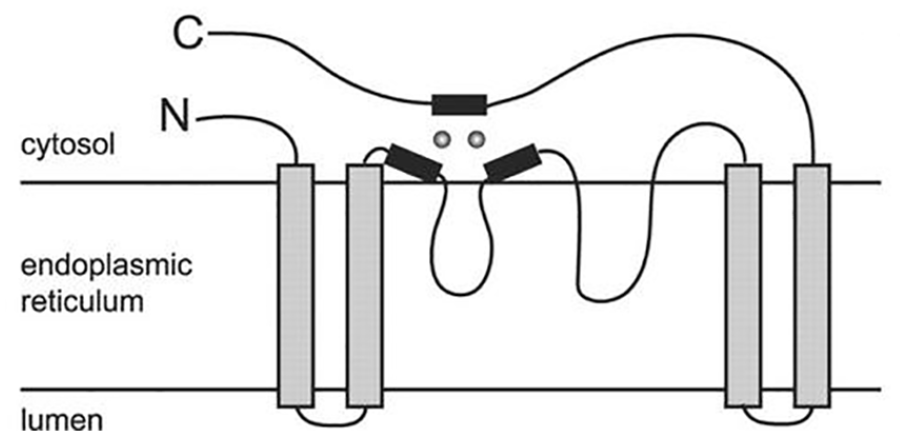
Topology of histidine and iron domains in fatty acid desaturase enzymes. 20 Black box: histidine residue; gray circle: iron atom.
Phylogenetic relationship of the R. trisperma FAD2 with other plants
The alignment of the FAD2 nucleotide sequences was performed using ClustalW (MEGA 7.0). Phylogenetic trees were prepared using the neighbor-joining algorithm with a bootstrap value of 1000. A phylogenetic tree was used to determine the closeness level of R. trisperma FAD2 and the FAD2 of several Euphorbiaceae species. This study used G. hirsutum, Durio zibethinus, Theobroma cacao, and Herrania umbratica from the Malvaceae family as outgroups. The addition of outgroups aimed to obtain convincing information from a more closely related sequence. Based on the phylogenetic tree (Figure 4), there were 2 main groups: the Euphorbiaceae family and the Malvaceae family. The Euphorbiaceae group consisted of VfFAD2, VmFAD2, JcFAD2, HbFAD2, MeFAD2, TsFAD2, and RcFAD2. The Malvaceae group consisted of GhFAD2, DzFAD2, TcFAD2, and HuFAD2. The RtFAD2 sequence was closely related to VfFAD2 and VmFAD2. This was supported by a bootstrap value of 100. The NCBI database shows that RtFAD2, VfFAD2, VmFAD2, JcFAD2, HbFAD2, and MeFAD2 belong to the subfamily Crotonoideae. 49 According to the phylogenetic tree, the FAD2 of the 6 species is more closely related than those of TsFAD2 (subfamily Hippomaneae) and RcFAD2 (subfamily Acalypheae).

RtFAD2 phylogenetic tree with several species of Euphorbiaceae. VfFAD2: V. fordii FAD2; VmFAD2: V. montana FAD2; RtFAD2: R. trisperma FAD2; JcFAD2: J. curcas FAD2; HbFAD2: H. brasiliensis FAD2; MeFAD2: M. esculenta FAD2; TsFAD2: T. sebifera FAD2; RcFAD2: R. communis FAD2; GhFAD2: G. hirsutum FAD2; DzFAD2: D. zibethinus FAD2; TcFAD2: T. cacao FAD2; HuFAD2: H. umbratica FAD2.
In silico analysis of FAD2 subcellular localization
Prediction of subcellular location can be performed by direct analysis using protein sequences. 50 Proteins have an intrinsic signal that has an essential function in targeting proteins to cellular organelles (signal peptides). 51 A signal peptide is an N-terminal peptide consisting of 15 to 30 amino acids that are released during the protein translocation process when it crosses the membrane. 25 The RtFAD2 subcellular location prediction was performed using the TargetP 1.1 Server program. RtFAD2 showed a high level of similarity to VfFAD2. Therefore, in this study, the subcellular location was predicted by comparing RtFAD2 and VfFAD2 (AEE69021.1). This subcellular location prediction included the probability that the signal peptide travels to the chloroplast, mitochondria, or secretory pathway. The level of confidence in the prediction results is indicated by the reliability class (RC; 1-5), where the high confidence values range from 1 to 3, while a low level of confidence is indicated when the RC is in the range of 4 or 5. 25
Based on the prediction results, RtFAD2 has a signal peptide that leads to the secretory pathway. This is indicated by the secretory pathway value of 0.83 (RC = 3). The results of this prediction differed from those of VfFAD2 (Table 4). The test results showed that VfFAD2 did not have a signal peptide indicating a subcellular location in either the chloroplast, mitochondria, or secretory pathway. However, the prediction results indicated that VfFAD2 was present in other cellular organelles (other values = 0.753; RC = 3). This was probably due to the differences in nucleotide sequences between RtFAD2 and VfFAD2. In addition, the RtFAD2 sequence obtained in this study was still partial. This is supported by other studies showing the complete sequence of FAD2, Salvia hispanica, and Brassica juncea. Arabidopsis does not have signal peptides that go to chloroplasts or mitochondria.37,52
Subcellular localization of RtFAD2 and VfFAD2.

Abbreviations: cTP, chloroplast transit peptide; FAD2, fatty acid desaturase 2; mTP, mitochondrial targeting peptide; RC, reliability class; SP, secretory pathway.
Further in silico analysis was conducted to predict whether R. trisperma FAD2 is a transmembrane protein. Our prediction showed that the partial sequence of RtFAD2 encoded a protein with 3 transmembrane domains (Figure 5), whereas VfFAD2 had 4 transmembrane domains (Figure 6). The transmembrane domain of RtFAD2 was found in amino acids 21-43, 115-137, and 189-211; conversely, the transmembrane domain of VfFAD2 was found in the 55-77, 82-104, 178-200, and 247-269 amino acids. Our data were in accordance with a previous study that showed that the FAD2 protein is integrated with the membrane of the endoplasmic reticulum. It has also been stated that FAD2 of V. fordii expressed on Bright-Yellow Nicotiana tabacum cv (BY-2) cells is located in the endoplasmic reticulum. 19 FAD2 transmembrane domain analysis has also been performed in silico on Sesamum indicum (5 domains, amino acids 55-77, 82-104, 119-136, 176-198, 223-272) and B. juncea (4 domains at amino acids 55-77, 82-104, 176-198, and 253-275).19,52

Topological prediction of FAD2 transmembrane domain from Reutealis trisperma. FAD2 indicates fatty acid desaturase 2.
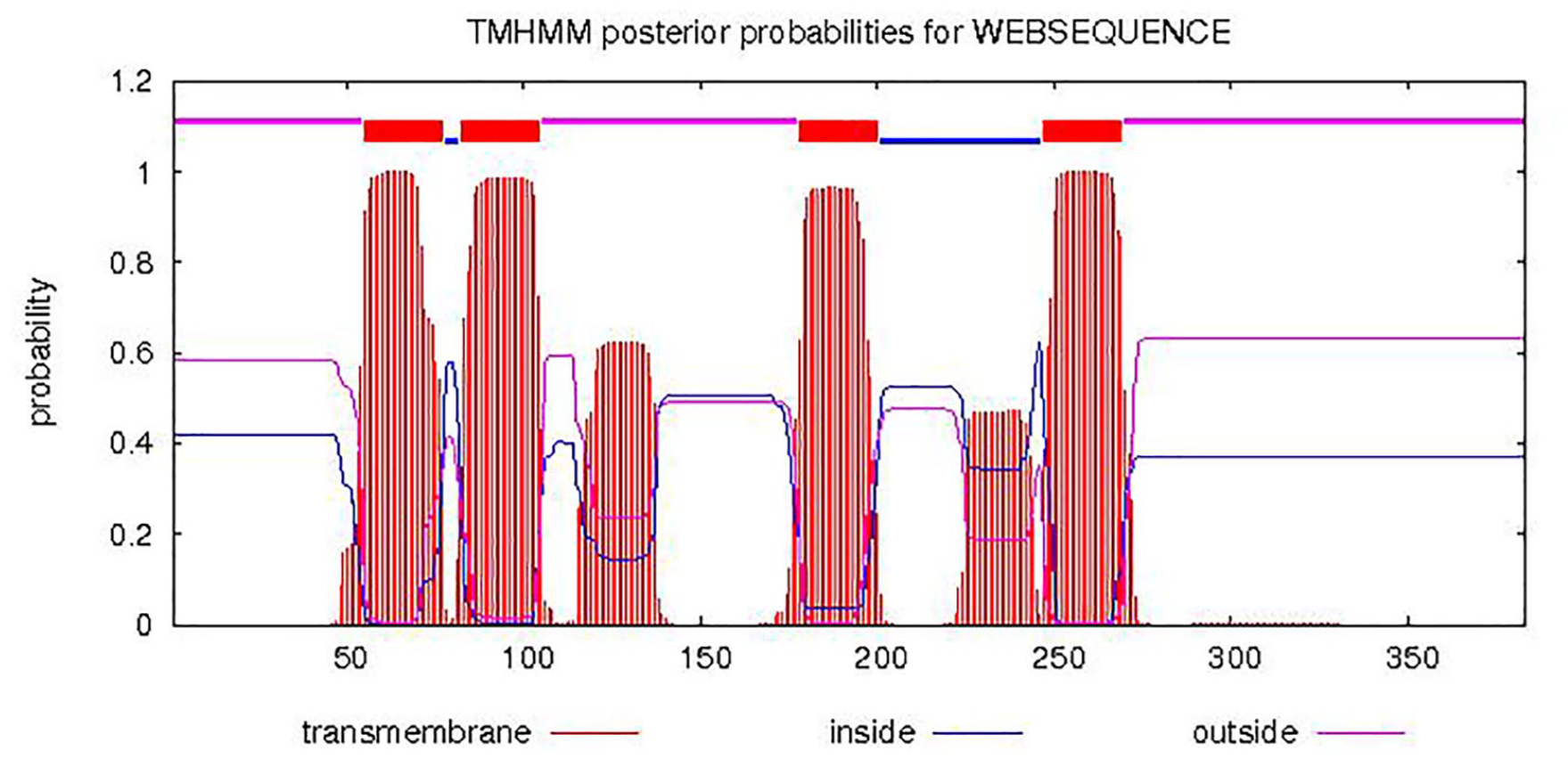
Topological prediction of FAD2 transmembrane domain from Vernicia fordii. FAD2 indicates fatty acid desaturase 2.
Fatty acid biosynthesis in plants occurs in plastids. The biosynthesis of these fatty acids is preceded by the initiation and elongation of the acyl chain. In this reaction, acetyl- and malonyl-CoA are required as precursors. Fatty acid synthesis in plastids only reaches a chain length of 18°C. The first desaturation stage of fatty acids is catalyzed by plastidial stearoyl-acyl carrier protein (ACP) desaturase. Plastidial fatty acid chain termination is catalyzed by acyl-ACP thioesterases. After termination, free fatty acids are activated to a CoA thioester by an acyl-coenzyme A synthetase (ACS) and exported from the plastid to the endoplasmic reticulum. Further modification of fatty acids occurs in the endoplasmic reticulum, including reactions of desaturation, hydroxylation, and elongation. 53 The FAD2 enzyme modifies the addition of double bonds (desaturation) of oleic acid to the C atom at position 12 (ω-6). 19 During seed development, acyl chain flow in the endoplasmic reticulum eventually initiates the esterification reaction. The fatty acid esterification reaction occurs at all 3 glycerol positions. The esterification reaction forms triacylglycerol, which is then stored in the seeds. 53
Conclusions
In this study, we isolated and characterized the partial cDNA of the FAD2 gene from R. trisperma. This gene is responsible for the formation of unsaturated fatty acids by catalyzing the introduction of a double bond in the hydrocarbon chain of fatty acids. We successfully isolated 923 bp of FAD2 cDNA sequences. In silico prediction demonstrated that this sequence encodes 260 amino acids and possesses an omega-6 FAD domain. In addition, our in silico study revealed that the FAD2 of R. trisperma is located in the endoplasmic reticulum membrane. The FAD2 fragment was also closely related to V. fordii (HM755946.1), another Euphorbiaceae plant. Finally, our data could pave the way for further research to improve the quality of R. Trisperma-based biodiesel through genetic modification of FAD2. Moreover, partial cDNA isolation of FAD2 might favor further studies on the expression dynamics of genes involved in the biodiesel biosynthesis of R. trisperma.
Footnotes
Acknowledgements
The authors gratefully acknowledge all members of the Laboratory of Plant Bioscience and Technology, Institut Teknologi Sepuluh Nopember (ITS). We also thank the Ministry of Research and Technology/National Research and Innovation Agency of the Republic of Indonesia, who has financed this project.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Ministry of Research and Technology/National Research and Innovation Agency of the Republic of Indonesia (1148/PKS/ITS/2020).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
N.J., D.E., and H.P. conceived and designed the experiments. I.P. and N.L.A.R. performed the bioinformatics analysis. N.J. and I.P. performed the molecular biology experiments. N.J., D.E., S.N., T.N., and H.P. supervised the experiments. N.J. and I.P. wrote the manuscript. All authors read and approved the final manuscript.