
Abstract
A new strain of the beta coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is solely responsible for the ongoing coronavirus disease 2019 (COVID-19) pandemic. Although several studies suggest that the spike protein of this virus interacts with the cell surface receptor, angiotensin-converting enzyme 2 (ACE2), and is subsequently cleaved by TMPRSS2 and FURIN to enter into the host cell, conclusive insight about the interaction pattern of the variants of these proteins is still lacking. Thus, in this study, we analyzed the functional conjugation among the spike protein, ACE2, TMPRSS2, and FURIN in viral pathogenesis as well as the effects of the mutations of the proteins through the implementation of several bioinformatics approaches. Analysis of the intermolecular interactions revealed that T27A (ACE2), G476S (receptor-binding domain [RBD] of the spike protein), C297T (TMPRSS2), and P812S (cleavage site for TMPRSS2) coding variants may render resistance in viral infection, whereas Q493L (RBD), S477I (RBD), P681R (cleavage site for FURIN), and P683W (cleavage site for FURIN) may lead to increase viral infection. Genotype-specific expression analysis predicted several genetic variants of ACE2 (rs2158082, rs2106806, rs4830971, and rs4830972), TMPRSS2 (rs458213, rs468444, rs4290734, and rs6517666), and FURIN (rs78164913 and rs79742014) that significantly alter their normal expression which might affect the viral spread. Furthermore, we also found that ACE2, TMPRSS2, and FURIN proteins are functionally co-related with each other, and several genes are highly co-expressed with them, which might be involved in viral pathogenesis. This study will thus help in future genomics and proteomics studies of SARS-CoV-2 and will provide an opportunity to understand the underlying molecular mechanism during SARS-CoV-2 pathogenesis.
Introduction
Evolution is a continuous and ongoing process driven by spontaneous mutations. While hosts develop defense mechanisms to counter pathogenicity, pathogens evolve to evade those defense systems inside the host. The recent pandemic manifestation of coronavirus disease 2019 (COVID-19) is one such instance for evolution and natural selection, where severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the sole perpetrator. 1 It is an RNA virus containing positive-sense, single-stranded RNA with a size of 26 to 32 kilobases (kb). Four major proteins named Spike (S), Envelope (E), Membrane (M), and Nucleocapsid (N) proteins are encoded by SARS-CoV-2 genome. This virus is named as coronavirus because of its crown shaped spikes on the outer membrane of the virus. 2 It is predicted that Chinese horseshoe bats may be the reservoirs for SARS-CoV2 and then transmitted from human to human. 3 Among almost 200 antigenically different types of respiratory illness-causing viruses, the Coronaviridae family is considered as one of the most common viruses that cause fatal respiratory infections. 4 They are grouped into four groups: alpha, beta, gamma, and delta viruses, with the Betacoronavirus family being the most pathogenic. 5 The seven forms of Betacoronavirus genus HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1, Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and the new SARS-CoV-2 are well known for their respiratory diseases caused in humans, and their ability to evolve from human immunity has made them more pathogenic to human hosts.5,6 SARS-CoV, MERS-CoV, and SARS-CoV-2 are possible Betacoronoviridae family members that are best known for their pandemic outbreaks of fatal respiratory infections in humans, while the other forms are linked to moderate respiratory illness. 7 During 2002-2003, SARS pandemic affected about 8500 persons killing 916 in 37 countries. 8 Similarly, by the end of January 2020, a total of 2519 laboratory-confirmed cases of MERS-CoV with 866 associated deaths were found in 27 countries. 9 On the contrary, as of June 10, 2021, more than 175 263 199 individuals are infected with SARS-CoV-2 with almost 3 779 147 deaths covering 220 countries globally.
Like other human coronaviruses, SARS-CoV-2 primarily affects the respiratory tract of humans. 10 The risk of transmission is increased by exposed mucous membranes and unprotected eyes as droplets and body fluid of infected patients can easily contaminate them. 11 Although contemporary research confirms that the virus interacts with the host protein angiotensin-converting enzyme 2 (ACE2) on the cell surface through its Spike (S) surface glycoproteins,12-14 recent findings suggest more than one entry system. 15 Entry of the virus is mediated by binding its surface unit S1, of the S protein to a host cellular receptor, allowing the virus to attach with the surface of target cells. Moreover, membrane fusion is depended on the cleavage of S protein by host cell proteases FURIN and TMPRSS2 at the site of S1/S2 and S2′, respectively, resulting in S protein activation (Supplemental Figure S1).
The unusual and new genetic makeup of SARS-CoV-2 has created a challenge in biological research. Bioinformatics play an increasingly large role in understanding the infectious diseases: from the pathogenesis, mechanisms, and the spread of diseases, to host immune responses.16,17 Meanwhile, in this difficult situation, bioinformatics emerged as one of the most important techniques for analyzing huge viral data. 18 Bioinformatics can help to discover susceptible genes and highlight the pathogenic processes that cause disease, allowing for the creation of effective therapies. 19 Bioinformatics is playing an immense role in the study of mutation annotation of viral and host proteins and their effect on host-pathogen interactions.16,20 Also, bioinformatics tools use various algorithms to evaluate the expression patterns of genes along with the effect of genetic variants on gene expression in different organs that allow identifying the host-pathogen interaction in a particular organ. As the use of experimental techniques is frequently related to the high proportion of both false-negative and false-positive predictions, computational approaches here play an important role to predict significant protein-protein interaction (PPI) in a short time. 21 Therefore, the use of bioinformatics tools helps to provide solutions to urgent biological problem concerning SARS-CoV-2 infection.
We believe that the effect of SARS-CoV-2 widely depends on two aspects: (1) genomic mutations of SARS-CoV-2 spike protein, ACE2, TMPRSS2, and FURIN as they are critically required for infection, and (2) expression profile of ACE2, TMPRSS2, and FURIN in different organs throughout the body. As the combined action of ACE2, TMPRSS2, and FURIN is necessary for the binding and uptake of the viral genetic material inside the host cell, in this study, we aim to identify whether variations in ACE2, TMPRSS2, and FURIN lead to an altered pattern of entry by changing their binding affinity and expression profile, thus bestowing host susceptibility to the infection. Besides this, we also consider PPIs and co-expression analysis of ACE2, TMPRSS2, and FURIN as another focal point to identify the functionally associated proteins and genes in viral infection. Protein-protein interaction offers a systemic identification of virus and host protein involvement in viral infection as well as targeting cellular antiviral target for effective treatment. 22
Methods
Protein characterization
Identification of physicochemical properties of the targeted proteins ACE2, TMPRSS2, and FURIN was performed by utilizing ProtParam from the ExPASy server. 23 ProtParam can compute various physical and chemical properties for a protein stored in Swiss-Prot or TrEMBL or for a user-submitted protein sequence. We submitted the UniProtKB ID of our targeted proteins to the server as input. From the results, we took the information about the molecular weight, theoretical pI, estimated half-life, instability index (II), and grand average of hydropathicity (GRAVY) of our targeted proteins.
PPI prediction
Protein-protein interactions provide both physical relations as well as functional associations between proteins. We used the STRING v.11.0 24 database to identify the proteins functionally interact with ACE2, FURIN, and TMPRSS2. STRING uses the backbone of biological machinery of proteins to build a PPI network. This type of connectivity network between proteins helps to understand the full biological phenomenon behind the proteins. We submitted a list of these three ACE2, TMPRSS2, and FURIN proteins as input data and considered “Homo sapiens” as target organism. STRING then searched and sorted out the proteins that have direct (physical) and indirect (functional) interactions with the targeted proteins. We considered the data from text mining, experiments, databases, co-expression, neighborhood, gene fusion, and co-occurrence as interaction evidence to build the network edges.
Pathway and functional enrichment analysis
Pathway and functional enrichment analysis was performed by utilizing WikiPathway 25 and ShinyGO v0.61, 26 respectively, to identify the cumulative functional biological roles of ACE2, TMPRSS2, and FURIN along with their associated proteins. For functional enrichment analysis, GO-BP (biological process) term was considered to identify the functional biological roles of ACE2, TMPRSS2, and FURIN. For protein pathway and functional enrichment analysis, we provided the official names of these three ACE2, TMPRSS2, and FURIN proteins as input and the species was set as “Human.” The P-value cutoff [False Discovery Rate (FDR)] was set to .05 to extract the 30 most significant biological processes through ShinyGO.
Tissue-based genotype-specific expression
Tissue-based expression analysis of ACE2, TMPRSS2, and FURIN genes was done by using GTEx Portal. 27 This analysis facilitated the identification of the tissue localization of these genes as well as their level of expression. The GTEx Portal includes the genotype data of these genes from 948 donors (male and female; age 20-79) and 17 382 RNA-seq samples across 54 tissue sites and 2 cell lines. The donors were densely genotyped to identify the genetic variations within their genomes. This portal quantified the expression of these genes from the 54 tissue sites whose pathological status includes no abnormalities, pneumonia, fibrosis, diabetic, adenoma, gynecomastoid, infarction, sclerotic, cirrhosis, gastritis, hypertrophy, atelectasis, glomerulosclerosis, hypoxic, hyalinization, nephrosclerosis, dysplasia, congestion, and hepatitis. Log scale (log 10) was considered for calculating expression value based on Transcripts Per Million (TPM). As these ACE2, TMPRSS2, and TMPRSS2 proteins are essential for the entry of SARS-CoV-2 into the host cell, through this expression analysis we can identify the specific tissues that are susceptible to the viral infection. In addition, we evaluated the genotype-specific expression (GSE) of ACE2, TMPRSS2, and FURIN in different human tissues to identify genetic variants affecting the normal expression of these genes through cis-expression quantitative trait loci (cis-eQTL) analysis by utilizing the GTEx Portal (GTEx Analysis Release V8). Violin plots of the GSE were used to visualize the normalized gene expressions. We accounted P-value cutoff ⩽.05 for statistical significance and Normalized Effect Size (NES) ⩾0.5 or ⩽−0.5 for larger effect in the cis-eQTL analysis.
Co-expression analysis
Coexpedia 28 and SEEK 29 databases were used to identify the genes that are co-expressed with ACE2, TMPRSS2, and FURIN. Both of the tools are query-based search engine and use very large transcriptomic data including human data sets from many different microarray and high-throughput sequencing platforms to predict co-expression network, but the SEEK has a unique strength of predicting genes co-expressed with multiple genes and cross-platform analysis. Coexpedia was used to determine the genes that are individually co-expressed with ACE2, TMPRSS2, and FURIN, whereas SEEK was utilized to identify the genes that are co-expressed with all three genes ACE2, TMPRSS2, and FURIN. Besides, enrichment analysis of these co-expressed genes was also performed to understand the functional conjugation of co-expressed genes with ACE2, TMPRSS2, and FURIN in viral infection. Official gene symbol was provided as input for both of the databases.
Sequence and data retrieval
The amino acid sequence of SARS-CoV-2 spike protein (YP_009724390.1) from NCBI 30 and the amino acid sequences of human ACE2 (Q9BYF1), TMPRSS2 (O15393), and FURIN (P09958) from UniProt 31 were retrieved. We then collected the variants of the ACE2, TMPRSS2, and FURIN genes and their allele frequencies from the Ensembl Genome Browser 32 and gnomAD database, 33 respectively. We annotated the coding variants of ACE2 to identify the variants that are involved in binding with receptor-binding domain (RBD) and selected for further analysis. 34 Similarly, we annotated the coding variants of TMPRSS2 and FURIN to identify the variants that are involved in binding with TMPRSS2 and FURIN cleavage sites within the spike protein, respectively.35-37
Sequence analysis of spike protein of SARS-CoV-2
The amino acid sequence of SARS-CoV-2 spike protein (YP_009724390.1) was taken as the reference sequence to carry out the PSI-BLAST 38 program with the target set to 1000. Output sequences associated with SARS-CoV-2 spike protein were retrieved for further analysis. We then performed Multiple Sequence Alignment (MSA) of the SARS-CoV-2 spike proteins through the ClustalW algorithm by utilizing MEGA 7 39 software. The alignment was visualized by JALVIEW software to mark the variations of SARS-CoV-2 spike proteins within the RBD and cleavage sites for TMPRSS2 and FURIN. In addition, CNCB database 40 was explored to confirm our findings as well as to extract new missense spike protein mutations.
Interaction analysis of ACE2 and RBD of SARS-CoV-2 through homology-based protein-protein docking and binding energy estimation
The co-crystal structure of the spike protein (RBD) of SARS-CoV-2 complexed to human ACE2 is available in Protein Data Bank (PDB). 34 We considered the PDB ID: 6LZG for its non-chimeric nature of the spike protein and resolution at a lower wavelength (2.5 Å). By taking this structure as a template, we built the complex of spike protein with the human ACE2 protein. To extract the RBD of spike protein and binding region of ACE2, the template sequences of the receptor (ACE2) and the ligand (RBD) were aligned locally with the target sequences using the Water program from the EMBOSS package. 41 Then, the SWISS-MODEL server 42 was utilized to model the complex of spike protein (RBD) and ACE2. Different three-dimensional (3D) structures of RBD-human ACE2 complex, each consisting of one of the identified RBD variants, were modeled by using the “Mutation tool” from Swiss PDB viewer. 43 Similarly, we modeled the 3D structures of RBD-human ACE2 complexes, each consisting of one of the identified ACE2 variants. Intermolecular hydrogen bonds, electrostatic, and hydrophobic interactions between RBD (native and mutants) and human ACE2 (native and mutants) were monitored using Discovery Studio and LigPlot+. To predict the binding affinity of the complexes, we used the PRODIGY 44 web server.
Protein molecular modeling
To construct the complete 3D structures of the spike protein of SARS-CoV-2, human TMPRSS2, and human FURIN, we used MODELLER (v9.23). 45 PSI-BLAST algorithm was carried out to find the appropriate template for structural construction with the source database set as PDB. From the BLAST result, we picked the structures that show >40% similarity and identity for comparative homology modeling. In the case of each protein modeling, MODELLER was instructed to generate 10 models. The best model was chosen to have the lowest Discrete Optimized Protein Energy (DOPE) score and the highest GA341 score. Then, the structural assessment was carried out by the Ramachandran Plot analysis through the RAMPAGE server. 46
Interaction analysis of TMPRSS2 and FURIN with spike protein of SARS-CoV-2 through protein-protein docking and binding energy estimation
We used the HADDOCK 2.4 server 47 to perform the protein-protein docking to figure out the intermolecular interactions of the TMPRSS2 and FURIN with spike protein of SARS-CoV-2 along with their binding affinity. For spike protein-TMPRSS2 docking, the modeled 3D structure of spike protein was submitted as molecule 1 and modeled 3D structure of TMPRSS2 as molecule 2. As K814 and R815 residues of spike protein act as the cleavage site for TMPRSS2, 48 we picked these two as active residues. Besides, the catalytically active H296, D345, and D435 residues were selected as active residues for TMPRSS2. 35 Similarly, for spike protein-FURIN docking, the modeled 3D structure of spike protein was submitted as molecule 1 and modeled 3D structure of FURIN as molecule 2. The P681, R682, R683, A684, and R685 residues were considered as active residues for spike protein, as they acted as the FURIN cleavage site. 48 On the contrary, R185, M189, D191, N192, R193, E229, V231, G230, D233, D259, K261, R298, W328, and Q346 residues were selected as active residues for FURIN.36,37 The best-docked complexes were chosen according to the HADDOCK and Z score. Different docked 3D structures of spike protein-human TMPRSS2 and spike protein-FURIN, each consisting of one of the identified variants of spike protein within the TMPRSS2 and FURIN cleavage sites, respectively, were modeled by using “Mutation tool” from Swiss PDB viewer. Similarly, TMPRSS2 and FURIN variants containing docked 3D structures were also created. Intermolecular hydrogen bonds, electrostatic, and hydrophobic interactions between spike protein (native and mutants) and human TMPRSS2 (native and mutants) were monitored using Discovery Studio and LigPlot+. Likewise, we also analyzed the intermolecular hydrogen bonds, electrostatic, and hydrophobic interactions between spike protein (native and mutants) and human FURIN (native and mutants). To predict the binding affinity of the complexes, the PRODIGY web server was utilized.
Overview of the methods of the study is briefly presented in a schematic diagram (Figure 1).

Schematic diagram summarizing the methods.
Results
Protein characterization
ProtParam server showed that the ACE2 precursor is 805 amino acids long, where 1 to 17 and 18 to 805 amino acids serve as a signal peptide and the active ACE2, respectively. The molecular weight and theoretical isoelectric point (pI) of ACE2 were predicted as 90 745 Da and 5.36, respectively. The half-life of ACE2 was estimated at 0.8 h (mammalian reticulocytes, in vitro). The instability index (II) of ACE2 was computed to be 39.40 that indicates the protein’s stability. The average hydropathicity (GRAVY) was predicted as −0.415. Amino acids from 18 to 740 were identified as the extracellular part of ACE2, which is crucial for binding with SARS-CoV-2 spike protein. More specifically, 30 to 41, 82 to 84, and 353 to 357 amino acids are essential for interaction. The amino acids that range from 697 to 716 were predicted as essential for cleavage by TMPRSS11D and TMPRSS2. TMPRSS2 is a transmembrane protease enzyme of 492 amino acids containing two chains, one is non-catalytic (1-255 amino acids) and the other is catalytically active (256-492 amino acids) that is essential for cleaving SARS-CoV-2 spike protein. The molecular weight and theoretical pI of the catalytic domain were predicted as 26 224.10 Da and 7.08, respectively. The estimated half-life of the catalytic domain was 20 h (mammalian reticulocytes, in vitro). The catalytic domain was predicted as stable with the instability index (II) value of 30.33. The average hydropathicity (GRAVY) was −0.106.
FURIN is a protease enzyme of 794 amino acids containing a peptidase S8 domain (121-435) and acts as a protease enzyme. Molecular weight and theoretical pI of peptidase S8 domain were identified as 33 539.80 Da and 5.29, respectively. The estimated half-life was 0.8 h (mammalian reticulocytes, in vitro). The average hydropathicity (GRAVY) was −0.420. The instability index (II) was computed to be 18.68 that means the domain is stable.
PPI analysis
Protein-protein interactions revealed that our queried proteins ACE2, FURIN, and TMPRSS2 have neighborhood interactions among themselves (Figure 2A) with two edges, where PPI enrichment P-value is .000298 (<.001; Supplemental Table S1). Moreover, ANTXR1, ANTXR2, MMP14, BACE1, MME, REN, AGT, TGFB, NOTCH1, and COL23A1 proteins were predicted as functional partners (first shell of inter-actors) having significant interactions with three of our targeted proteins (Figure 2B). The interaction network of total 23 proteins showed 61 edges, where the PPI enrichment P-value is 7.97E–08 (Supplemental Table S2).
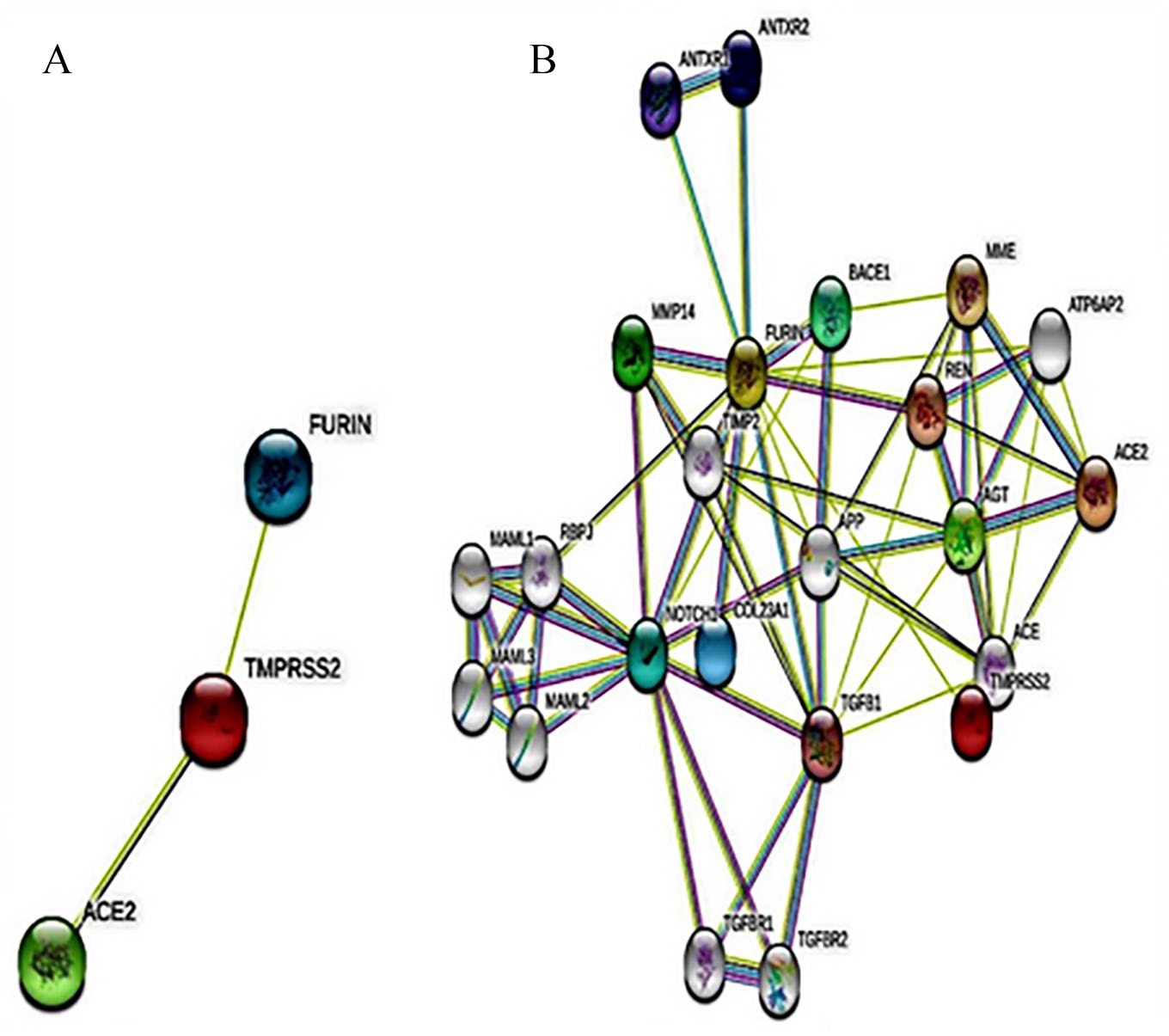
PPI network of ACE2, FURIN, and TMPRSS2. (A) Interactions among the three target proteins. (B) Interactions among 20 neighbor proteins along with the target proteins. Here, light pink and light blue edges indicate known interactions from curated databases and experimental determination, respectively; green, red, and blue edges indicate predicted interactions of gene neighborhood, gene fusions, and gene co-occurrence, respectively. The colored nodes indicate query proteins and the first shell of inter-actors and white nodes indicate the second shell of inter-actors. PPI indicates protein-protein interaction.
Functional enrichment analysis
Identification of association of ACE2, TMPRSS2, and FURIN in viral infection pathway through WikiPathway revealed that the surface glycoprotein S is cleaved by the host protease FURIN TMPRSS2 which produces S1 and S2 subunits. The S1 subunit contains the RBD, which directly binds to the ACE2. After binding with the ACE2 peptidase domain, another cleavage site on S2 is exposed and cleaved by the other host protease TMPRSS2 FURIN, thereby producing S2′ subunit, which is crucial for membrane fusion. These interactions help the virion to regulate the expression of the host proteins that lead to pathogenesis.25,48 We performed enrichment analysis to understand the functional role of these host proteins in the pathway of viral infection through ShinyGO v0.61. This analysis showed that ACE2, TMPRSS2, and FURIN are involved in peptidase activity, protein processing, protein maturation, viral life cycle, and viral process. Specifically, ACE2 and FURIN are involved in the receptor biosynthetic process and receptor metabolic process. FURIN and TMPRSS2, on the contrary, are involved in the activity of serine-type peptidase, endopeptidase, and viral entry into the host cell. The biological processes of ACE2, TMPRSS2, and FURIN are shown in Figure 3. Pathway and enrichment analysis predicted that cumulative action of ACE2, TMPRSS2, and FURIN is needed to perform many of these biological processes in SARS-CoV-2 infection (Supplemental Table S3).

Representation of the biological processes involving ACE2, TMPRSS2, and FURIN after performing enrichment analysis by ShinyGO server.
Tissue-based gene and protein expression
Gene expression analysis from GTEx Portal showed that the ACE2 gene had a relatively higher level of expression in the small intestine, testis, breast, ovary, and thyroid. On the contrary, the adrenal gland, pituitary, uterus, bladder, liver, spleen, and blood showed the lowest ACE2 expression. Angiotensin-converting enzyme 2 expression was found to moderate in the lung, minor salivary gland, fallopian tube, nerve, pancreas, prostate, stomach, and vagina (Supplemental Figure S2a). Similarly, the TMPRSS2 gene was found to express at a greater level in the prostate, lung, stomach, pancreas, bladder, breast, thyroid, and small intestine, while it showed the lowest expression in muscle, nerve, ovary, spleen, blood, and uterus. Besides, the fallopian tube, liver, minor salivary gland, and vagina showed a medium expression level of TMPRSS2 (Supplemental Figure S2b). FURIN gene had relatively higher expression in breast, liver, lung, minor salivary gland, bladder, muscle, nerve, pancreas, pituitary, prostate, spleen, thyroid, and blood, while testis, bladder, fallopian tube, ovary, small intestine, stomach, uterus, and vagina had medium level of expression of FURIN gene (Supplemental Figure S2c).
We then performed GSE of these genes to investigate whether the genetic variants of these genes change their expression level. Genotype-specific expression analysis showed a total of 305, 22, and 78 genetic variants (P value ⩽0.05 and NES ⩾0.5 or ⩽−0.5) that effect normal expression level of ACE2, TMPRSS2, and FURIN, respectively (Supplemental Table S4). Among 305 variants of ACE2, we did not notice any genetic variants which affect its expression in lung tissue rather identified four significant variants rs2158082 (G > A), rs2106806 (A > G), rs4830971 (T > C), and rs4830972 (G > A) having an effect on its expression in brain tissue. Variants rs2158082 (P = 3.60E–16, NES = 0.64), rs2106806 (P = 3.60E–16, NES = 0.64), rs4830971 (P = 3.60E–16, NES = 0.64), and rs4830972 (P = 3.60E–16, NES = 0.64) make the ACE2 expression level higher than normal in brain (cortex) tissue. All of these four changes were found to occur in X chromosome. Among 22 variants, we identified 4 significant TMPRSS2 variants rs458213 (T > A), rs468444 (G > A), rs4290734 (A > G), and rs6517666 (C > A) having an effect on its expression in lung tissue.
Variants rs458213 (P = 1.70E–08, NES = 0.59), rs468444 (P = 7.00E–08, NES = 0.63), and rs4290734 (P = 8.30E–10, NES = 0.69) make the TMPRSS2 expression level higher than normal in lung tissue, whereas rs6517666 (P = 3.90E–08, NES = −0.53) makes the TMPRSS2 expression level lower than normal. All of these four changes were found to occur in chromosome 21.
In the same way, we recognized two significant variants rs78164913 (T > G) and rs79742014 (C > T) of FURIN having an effect on its expression in lung tissue. These two changes rs78164913 (P = 1.30E–31, NES = −1.6) and rs79742014 (P = 3.30E–26, NES = −1.4) were found to occur in chromosome 15 and make the FURIN expression level lower than normal in lung tissue. All the above-mentioned GSE is shown in Figure 4.

Violin plots of genotype-specific expression of ACE2, TMPRSS2, and FURIN in different human tissues. The teal region indicates the density distribution of the samples in each genotype. In each violin plot, the first and (second and third) teal region indicates the reference and alternative allele type, respectively. The white line in the box plot (black) shows the median value of the expression of each genotype.
Co-expression analysis
Co-expression analysis of ACE2, TMPRSS2, and FURIN by Coexpedia showed that 146, 373, and 60 genes are co-expressed with ACE2, TMPRSS2, and FURIN, respectively (Supplemental Tables S5-S7). GLYAT, DDC, SLC13A1, TMEM27, and FMO1 genes are highly co-expressed with ACE2 having scores (sum of log-likelihood scores from all co-expression links) 49 8.97, 8.24, 6.46, 5.19, and 5.05, respectively. According to these scores, GLYAT acts as a hub gene. Functional enrichment analysis of these genes showed that they are involved in biological processes such as type I interferon signaling pathway, negative regulation of viral genome replication, response to virus, drug catabolic process, chemokine-mediated signaling pathway, receptor biosynthetic process, T cell chemotaxis, positive regulation of monocyte chemotaxis, B cell activation involved in immune response, positive regulation of B cell apoptotic process, and so on. Similarly, C1orf116, SLC44A4, TMEM125, HOXB13, and KLK2 are highly co-expressed with TMPRSS2 with the values of 12.35, 8.57, 7.12, 5.90, and 5.43, respectively. Following the scores, C1orf116 acts as a hub gene. Biological processes such as cellular response to hypoxia, negative regulation of cell adhesion, drug catabolic process, platelet degranulation, activation of protein kinase activity, oxidation-reduction process, cell-cell junction organization, endocytosis, regulation of gene expression, and so many functions are associated with these co-expressed genes of TMPRSS2. Among the 60 genes co-expressed alongside FURIN, a higher level of co-expression was observed for NPPA, MAPK8IP2, AMN, SOX15, and LHB with the scores of 3.10, 2.70, 2.67, 2.54, and 2.53, respectively. Based on scores, NPPA was found as a hub gene. These co-expressed genes are involved in negative regulation by host of viral release from the host cell, viral protein processing, negative regulation by virus of viral protein levels in the host cell, evasion or tolerance of host defenses by the virus, positive regulation by host of viral genome replication, positive regulation by host of viral release from the host cell, modulation by virus of host morphology or physiology, and so many.
Moreover, by utilizing SEEK tool, we identified 10 genes (FUT2, MUC3A, B3GNT3, C1ORF116, ELF3, FUT3, PCDH1, MYH14, PRSS8, and LAD1) that are commonly co-expressed with ACE2, TMPRSS2, and FURIN (Supplemental Figure S3). Among them, functional interactions exist between FUT2, FUT3, MUC3A, and B3GNT3 (Supplemental Figure S4). FUT2 and FUT3 interact with each other and with B3GNT3 through glycosyl transferase domain. FUT2 and FUT3 are involved in fucosylation, fucose metabolic process, protein glycosylation, and macromolecule glycosylation, whereas MUC3A and B3GNT3 are involved in macromolecule glycosylation and protein glycosylation.
Data retrieval and annotation
A total of 223 missense variants of ACE2 were identified from gnomAD and Ensembl Genome Browser (Supplemental Table S8). Among them, we found seven variants, which are at critical positions that are essential for the binding of ACE2 with the viral RBD of spike protein (Supplemental Table S9). Allele frequencies of these seven variants ranged from 3.44E–5 (rs143936283) to 1.09E–5 (rs781255386). Similarly, out of 294, we identified 3 missense variants of TMPRSS2 at critical positions that are essential for the binding with the viral TMPRSS2 cleavage site of spike protein (Supplemental Tables S10 and S11). Allele frequencies of these three variants of TMPRSS2 ranged from 8.64E–6 (rs906113408) to 3.98E–6 (rs867186402). Moreover, we also identified 4 missense variants of FURIN out of 366, which are at critical positions that are essential for the binding with the viral FURIN cleavage site of spike protein (Supplemental Tables S12 and S13). Allele frequencies of these four variants of FURIN ranged from 8.03E–6 (rs749858583) to 3.99E–6 (rs1347562753).
Sequence analysis
Among the 1000 output sequences from the PSI-BLAST result, 835 sequences were of SARS-CoV-2 spike protein, which was retrieved for analysis. From Multiple Sequence Alignment of 835 spike protein sequences and CNCB database, we identified 61 missense mutations that are in critical position for binding with ACE2, TMPRSS2, and FURIN. Among 61 missense mutations, 27, 14, and 20 mutations were found within the RBD region (P333-P527), close to TMPRSS2 cleavage site (K814-R815), and within FURIN cleavage site (P681-R685), respectively. Findings from CNCB database are displayed in Supplemental Table S14.
Interaction and binding energy analysis of ACE2 and RBD of SARS-CoV-2
The modeled complex structure of wild-type SARS-CoV-2 spike protein (RBD) with wild-type ACE2 accentuated the interaction points of both the proteins. These interactions were also reported in our template resolved structure of the SARS-CoV-2 spike protein (RBD)-ACE2 complex (PDB ID: 6LZG). A representation of the comparison of these interactions for different ACE2 variants is shown in Supplemental Table S15. We also enlisted the comparison of these interactions for different RBD mutations in Supplemental Table S16.
Intermolecular interaction analysis between the wild-type SARS-CoV-2 spike protein (RBD) and wild-type ACE2 revealed that the K31, D30, S19, Q24, Y41, K353, D38, Q42, Y83, E35, E37, H34, and Y84 residues of the wild-type ACE2 form hydrogen bonds with the E484, K417, A475, N487, T500, (G496 and G502), (Y449 and G496), (Y449 and Q498), N487, Q493, Y505, Y453, and F486 residues of the wild-type SARS-CoV-2 spike protein, respectively. On the contrary, the H34, Y83, (K353 and G354), M82, K31, and K353 residues of the wild-type ACE2 hydrophobically interact with the L455, F486, Y05, F486, Y489, and Y505 residues of the wild-type SARS-CoV-2 spike protein, respectively. Moreover, the K31 and D30 residues of the wild-type ACE2 establish electrostatic interactions with the E484 and K417 residues of the wild-type SARS-CoV-2 spike protein, respectively. Besides, the overall binding energy between wild-type ACE2 and wild-type SARS-CoV-2 spike protein was calculated to be −12.5 kcal/mol.
We analyzed the effect of seven missense variants of ACE2 on the binding interaction of ACE2 with SARS-CoV-2 spike protein (RBD) and observed that E35K, E37K, M82I, E329G, and D355N variants of ACE2 have no significant alteration in binding interaction. S19P variant of ACE2 showed relatively less binding affinity (−12.1 kcal/mol) compared with wild-type complex. Also, the hydrogen bond between S19 and A475 was found missing that was present in the wild-type complex. Moreover, the mutant showed 4 polar-polar, 22 polar-apolar, and 10 apolar-apolar interfacial contacts (ICs), whereas the wild-type complex showed 5 polar-polar, 23 polar-apolar, and 8 apolar-apolar ICs. The most prominent alteration in binding energy was found for the mutant T27A with the binding energy of −11.6 kcal/mol. In addition, A27 participates in the formation of an alkyl bond with A475, which was not present in the wild-type complex (Figure 5). Likewise, the mutant T27A showed 19 polar-apolar and 12 apolar-apolar ICs, whereas the wild-type complex showed 23 polar-apolar and 8 apolar-apolar ICs.

Intermolecular interactions of ACE2 (T27A) with SARS-CoV-2 RBD. Here, the residues of the ACE2 and the spike protein (RBD) are marked as A and B, respectively. ACE2 indicates angiotensin-converting enzyme 2; RBD, receptor-binding domain; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
We also analyzed the effect of 27 missense variants of SARS-CoV-2 spike protein (RBD) on the binding interaction of spike protein with ACE2 and observed that L452Q, T478K, L455F, F456L, S459F, A475V, N439K, L452R, T470N, E484D, E484A, E484K, E484Q, F486L, S494P, S494L, N501T, N501Y, F490L, F490S, S477N, S477T, E471D, and E471Q variants of the SARS-CoV-2 spike protein (RBD) showed almost similar interactions when compared with the wild-type complex along with having almost similar binding affinity (−12.5 kcal/mol). On the contrary, relatively higher binding energy (−12.9 kcal/mol) and absence of hydrogen bond between Q493 and E35 were observed for Q493L when compared with wild-type complex (Supplemental Figure S5). Moreover, the mutant Q493L showed 7 charged-polar and 24 charged-apolar ICs, whereas the wild-type complex showed 10 charged-polar and 20 charged-apolar ICs. The most prominent alteration in binding energy was found for the mutant G476S with the binding energy of −11.7 kcal/mol. In addition, alteration in binding interactions was not observed (Supplemental Figure S6). Moreover, the mutant G476S showed 7 polar-polar and 21 polar-apolar ICs, whereas the wild-type complex showed 5 polar-polar and 23 polar-apolar ICs. The highest prominent change in binding energy was noticed for mutant S477I and that is −13.3 kcal/mol along with no alteration in intermolecular interactions (Figure 6).

Intermolecular interactions of ACE2 with SARS-CoV-2 RBD (S477I). Here, the residues of the ACE2 and the spike protein (RBD) are marked as A and B, respectively. ACE2 indicates angiotensin-converting enzyme 2; RBD, receptor-binding domain; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Protein molecular modeling through MODELLER v9.23
PDB ID 6VSB, 6VXX, and 6VYB showed 99.59% identity with the target spike protein and were used as templates for modeling the complete spike protein. Similarly, PDB ID 5TJZ, 2ANY, and 2ANW showed 42.56%, 42.5%, and 42.5% identity with the target TMPRSS2 protein, respectively, and were used as templates for modeling the complete TMPRSS2 protein. Moreover, PDB ID 4OMC, 1P8J, and 4Z2A showed 99.36%, 97.88%, and 99.57% identity with the target FURIN, respectively, and were used as templates for modeling the FURIN protein. Based on the lowest DOPE score and the highest GA341 score, we selected the best model and selected the 5th, 9th, and 2nd models of the spike protein, TMPRSS2, and FURIN, respectively, from the summary of the models for the quality assessment. Through Ramachandran Plot analysis, we found that the model of spike protein, TMPRSS2, and FURIN had 92.7%, 84.1%, and 91.5% amino acids in the favored region, respectively (Supplemental Figure S7). Hence, these models were considered to be good and reliable for further analyses.
Interaction analysis of TMPRSS2 and FURIN with spike protein of SARS-CoV-2 through protein-protein docking and binding energy estimation
To monitor the binding interactions of the TMPRSS2 and FURIN with the viral spike protein, we performed molecular docking by HADDOCK 2.4 server. A representation of the comparison of binding interactions for different TMPRSS2 variants as well as for different spike protein mutations in TMPRSS2 cleavage sites is shown in Supplemental Tables S17 and S18, respectively. Similarly, comparison of binding interactions for different FURIN variants as well as for different spike protein mutations in FURIN cleavage sites is shown in Supplemental Tables S19 and S20, respectively.
From the intermolecular interaction analysis between the wild-type SARS-CoV-2 spike protein (TMPRSS2 cleavage sites) and wild-type TMPRSS2, we observed that the K811, D796, D936, S810, S929, K933, S939, D808, Q836, and P793 residues of the wild-type spike protein participated in the formation of a hydrogen bond with the E119, K80, (H296 and T309), D345, N304, (N303 and P301), H296, Y326, E329, and T78 residues of the wild-type TMPRSS2 protein, respectively. Besides, K814, F817, P793, K933, and Y837 residues of the wild-type spike protein formed hydrophobic interactions with the Y326, F311, V76, P301, and V331 residues of the wild-type TMPRSS2, respectively. Besides, the K811, D796, and D936 residues of the wild-type spike protein have been found to have an electrostatic bond with the E119, K80, and H296 residues of the wild-type TMPRSS2, respectively. The overall binding energy between wild-type spike protein and the wild-type TMPRSS2 protein was predicted as −11.2 kcal/mol.
We analyzed the effect of three missense variants of TMPRSS2 on the binding interaction of TMPRSS2 with SARS-CoV-2 spike protein (TMPRSS2 cleavage sites). For the D435N and A347T mutants of TMPRSS2, there was no change in binding affinity as well as in binding interactions. The most significant change was noticed for the mutant C297T in binding affinity with a score of −10.8 kcal/mol. Moreover, for the mutant C297T, the protein complex showed 8 polar-polar and 23 polar-apolar ICs, whereas the wild-type complex showed 7 polar-polar and 24 polar-apolar ICs. In addition, alteration in binding interactions was not observed for C297T (Supplemental Figure S8).
Afterward, we analyzed the effect of 14 missense variants of SARS-CoV-2 spike protein (TMPRSS2 cleavage sites) on the binding interaction of TMPRSS2 with SARS-CoV-2 spike protein (TMPRSS2 cleavage sites). Out of P812L, P812S, S813I, S813G, K814R, K814T, K814Q, K814E, K814M, K814N, K815S, K815M, K815K, and K815G mutants, only the P812S mutant of the spike protein revealed a significant change in binding affinity (−10.8 kcal/mol). Eight polar-polar and 23 polar-apolar ICs were also noticed for mutant P812S as well as no alteration was observed in intermolecular interactions (Supplemental Figure S9).
On the contrary, intermolecular interaction analysis between the wild-type SARS-CoV-2 spike protein (FURIN cleavage sites) and FURIN exhibited that the R214, R682, R683, R21, N211, G219, K278, N280, N282, N606, N679, S929, K933, Q1071, E1072, S689, Q690, T604, P681, and D215 residues of the wild-type spike protein were found to form hydrogen bonds with the E697, (E230 and G229), (D191 and M189), (P695, R693, and L694), K463, Q594, G527, G527, D526, K88, (D191 and N192), D177, N176, Y186, (Y186 and R357), K588, K588, T589, D228, and R703 residues of FURIN, respectively. In addition, R21, K182, and R214 residues of the wild-type spike protein showed hydrophobic interactions with the K469, L694, L704, and P696 of the wild-type FURIN, respectively. The R214, R682, R683, R685, K933, E1072, D215, and W258 residues of wild-type spike protein and the E697, E230, D191, E257, D177, E230, R357, R703, and K469 residues of FURIN showed electrostatic interactions between them, respectively. The binding affinity between the wild-type spike protein and FURIN was −13.2 kcal/mol.
We analyzed the effect of four missense variants of FURIN on the binding interaction of FURIN with SARS-CoV-2 spike protein (FURIN cleavage sites). The R298W mutant of FURIN showed the same binding affinity and interaction as the wild type. Whereas the mutant E230K also showed the same binding affinity, but hydrogen bond between R682-E230 and K933-E230 was missing when compared with the wild-type complex. For the mutant R193T, there was no change in binding interactions but showed slightly lower binding affinity (−13.0 kcal/mol) compared with the wild-type complex. Moreover, for the mutant R193T, the protein complex was found to have 39 charged-polar and 20 polar-polar ICs, whereas the wild-type complex showed 40 charged-polar and 19 polar-polar ICs. Interestingly, the mutant R185W showed a slightly more binding affinity (−13.5 kcal/mol). Also, for this mutant, a new pi-alkyl bond was formed between P681 and W185 that was absent in the wild type. In addition, for the mutant R185W, the protein complex was found with the 37 charged-polar, 29 charged-apolar, 34 polar-apolar, and 17 apolar-apolar ICs, whereas the wild-type complex showed 40 charged-polar, 31 charged-apolar, 32 polar-apolar, and 15 apolar-apolar ICs.
Furthermore, we analyzed the effect of 20 missense variants of SARS-CoV-2 spike protein (FURIN cleavage sites) on the binding interaction of FURIN with SARS-CoV-2 spike protein (FURIN cleavage sites). No significant change was found in binding energy as well as in interactions for the spike protein mutants P681L, P681H, P681S, P681T, R682W, R682Q, R682L, R683Q, R683P, R683L, A684E, A684P, A684T, A684S, A684V, R685C, R685G, and R685S. In case of mutants P681R and R683W, slight higher binding affinity (−13.5 kcl/mol) was observed along with the formation of new two hydrogen bonds between P681-D177 and P681-D228.
Discussion
The entry of SARS-CoV-2 into the host cell essentially depends on the cell receptor ACE2 and two serine proteases named TMPRSS2 and FURIN. Since the beginning of the COVID-19 pandemic, many experiments explained the interactions between SARS-CoV-2 spike protein and ACE2 as well as their mutation profiling to identify the key residues and to understand the method of entry into the host cell. However, sufficient attention was absent for investigating the effects of mutations inside the RBD and cleavage sites for TMPRSS2 and FURIN on viral pathogenesis. Besides, the impact of natural coding variants of human ACE2, TMPRSS2, and FURIN in binding with the spike protein is also lacking. Moreover, GSE of ACE2, TMPRSS2, and FURIN also affect the viral infection and this issue is also being given less importance. Thus, in this study, we focus on the variation in the binding pattern of the spike protein (RBD and cleavage sites) with ACE2, TMPRSS2, and FURIN due to non-synonymous single nucleotide polymorphisms (nsSNPs). In addition to mutation analysis, we also focus on the interactions of ACE2, TMPRSS2, and FURIN proteins with other proteins; functional conjugation between themselves; tissue-based GSE; and co-expression, and finally, we suggest some of the already existing drugs to combat COVID-19.
Neighborhood relationships among ACE2, TMPRSS2, and FURIN proteins indicate potential functional conjugation among these proteins. The larger number of edges for a random set of proteins of similar size than expected is strong evidence that they have more interactions among themselves. These types of interactions reveal that these three proteins are biologically connected with each other and also functionally associated with ANTXR1, ANTXR2, MMP14, BACE1, MME, REN, AGT, TGFB, NOTCH1, and COL23A1. Besides ACE2, TMPRSS2, and FURIN, these proteins could be the potential therapeutic targets in SARS-CoV-2 infection. Here, ACE2 acts as a host receptor for SARS-CoV-2 RBD, whereas FURIN and TMPRSS2 are involved in proteolysis of S1/S2 and S2′ cleavage sites, respectively, and subsequent fusion of the virus with the host cell membrane. 50 Thus, these outcomes suggest that these three proteins are essentially responsible for COVID-19 infection. As SARS-CoV-2 predominantly targets the lung tissue, 51 we wished to observe whether the expression of the ACE2, TMPRSS2, and FURIN genes is limited to the lungs. We found that apart from the lung, these genes are effectively expressed in some other tissues like small intestine, prostate, testis, breast, thyroid, liver, stomach, pancreas, vagina, ovary, and uterus as well. Previous findings also reported that the expression of the ACE2, TMPRSS2, and FURIN genes is high in these tissues.52-54 This suggests that besides lung tissue, there is also a possibility of SARS-CoV-2 infection in other tissues too. Genotype-specific expression concludes that genetic variants rs2158082, rs2106806, rs4830971, and rs4830972 make higher expression of ACE2 in brain tissue and might produce several neurological complications during COVID-19 as previous study demonstrated that SARS-CoV-2 has the neuro-invasive potentiality. 55 Higher level of expression of TMPRSS2 in lung was found for TMPRSS2 variants (rs458213, rs468444, and rs4290734). In case of these variants, susceptibility of lung tissue to SARS-CoV-2 might increase. Lower level of expression of TMPRSS2 and FURIN was observed for TMPRSS2 variant rs6517666 and FURIN variants (rs78164913 and rs79742014), respectively. Variants which lower the expression of TMPRSS2 and FURIN in lung tissue might reduce the susceptibility to infection in this particular organ.
We hypothesized that genes other than ACE2, TMPRSS2, and FURIN might also be involved in SARS-CoV-2 infection. Co-expression analysis revealed that genes (GLYAT, DDC, SLC13A1, TMEM27, and FMO1) that are highly co-expressed with ACE2 may help to initiate antiviral immunity by positive regulation of type I interferon signaling pathway, chemokine-mediated signaling pathway, T cell chemotaxis, monocyte chemotaxis, B cell activation, and negative regulation of viral genome replication. Likewise, through positive regulation of cellular response to hypoxia, drug catabolic process, platelet degranulation, and negative regulation of cell adhesion, genes (C1orf116, SLC44A4, TMEM125, HOXB13, and KLK2) that are co-expressed with TMPRSS2 may establish a barrier against viral pathogenesis. Finally, genes co-expressed with FURIN (NPPA, MAPK8IP2, AMN, SOX15, and LHB) also attempt to antiviral activity by involving themselves in reducing viral protein levels in the host cell and positive regulation of viral release from the host cell. Surprisingly, some of these genes may also facilitate viral infection through evasion or tolerance of host defenses by virus, positive regulation of viral genome replication, and modulation of host morphology or physiology. Moreover, genes (FUT2, MUC3A, B3GNT3, and FUT3) commonly co-expressed with all of the three targeted genes are associated with fucosylation and glycosylation of viral proteins, which leads us to believe that these genes possibly facilitate viral pathogenesis.
We analyzed the effects of the 61, 7, 3, and 4 nsSNPs of the genes encoding the spike protein (RBD and cleavage sites), ACE2, TMPRSS2, and FURIN, respectively, to investigate the alteration in binding affinity and interactions. We observed that the T27A variant of ACE2 significantly lowered the binding affinity with the spike protein as a change was noticed in polar-apolar and apolar-apolar ICs as well as in intermolecular interactions. Besides this ACE2 variant, the G476S mutation within the RBD region significantly decreased the binding affinity with ACE2 by the change in polar-polar and polar-apolar ICs. Our findings suggest that these two variants may exert reduced binding affinity between SARS-CoV-2 RBD and ACE2. Both the T27A and G476S mutations have previously been demonstrated to be associated with reduced SARS-CoV-2 entry.56-59 In addition, Q493L and S477I mutants of spike protein change the ICs and importantly increase the binding affinity of spike protein (RBD) with ACE2. Q493L mutation has previously been predicted to be involved with increased stability of RBD. 60 Moreover, the C297T and P812S variants of the TMPRSS2 and its cleavage site within the spike protein, respectively, showed a notable reduction in binding affinity with the alteration of polar-polar and polar-apolar ICs. This suggests that the mutants C297T and P812S may lead to resistance in the cleaving of the spike protein by the TMPRSS2. On the contrary, no significant alteration in binding energy, as well as binding interactions for the selected mutants of FURIN, was observed. The mutants P681R and R683W within the FURIN cleavage site change the intermolecular interactions and slightly increase the binding of FURIN with spike protein that may facilitate the cleaving.
We are positive that this study will thus help in understanding the underlying molecular events during viral infection due to genomic variations which might accelerate the future research on SARS-CoV-2 mutations.
Conclusion
In this study, we investigated the effects of the mutations on binding affinity and intermolecular interactions of SARS-CoV-2 spike protein with ACE2, TMPRSS2, and FURIN. Our analysis predicted several mutations that have the ability to significantly affect the host-virus interaction. Moreover, expression analysis suggests the possibility of viral infection in other organs along with the lung. Genotype-specific expression also predicted some genetic variants that notably change the expression of ACE2, TMPRSS2, and FURIN in different human tissues. Besides, we identified some genes that are highly co-expressed with ACE2, TMPRSS2, and FURIN, which may build up a barrier against viral infection. The results from this study will pave the way for future genomics and proteomics studies of SARS-CoV-2 and provide valuable insights for the development of new effective and safe drug/vaccine to treat COVID-19.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-1-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-10-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-10-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-11-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-11-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-12-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-12-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-2-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-2-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-3-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-3-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-4-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-4-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-5-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-5-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-6-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-6-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-7-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-7-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-8-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-8-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-9-bbi-10.1177_11779322211054684 – Supplemental material for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach
Supplemental material, sj-docx-9-bbi-10.1177_11779322211054684 for Prediction of the Effects of Variants and Differential Expression of Key Host Genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 Pathogenesis: An In Silico Approach by Md. Shahadat Hossain, Mahafujul Islam Quadery Tonmoy, Atqiya Fariha, Md. Sajedul Islam, Arpita Singha Roy, Md. Nur Islam, Kumkum Kar, Mohammad Rahanur Alam and Md. Mizanur Rahaman in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
M.S.H. acknowledges the Department of Biotechnology and Genetic Engineering, Noakhali Science and Technology University, for providing the research work supports. M.S.H. also acknowledges the Research Cell, Noakhali Science and Technology University and R&D, Ministry of Science and Technology, for research support. We also thank Mr. Tamer Ahamed, Norwich Medical School, University of East Anglia, for his English grammar correction throughout the paper.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
M.S.H. and M.M.R. conceived and designed this study. M.S.H., M.I.Q.T., A.F., A.S.R., and M.N.I. experimented and analyzed the data. M.S.I., M.I.Q.T., A.F., K.K., and M.R.A. wrote the manuscript. M.S.H., M.S.I., and M.M.R. made critical revisions. All authors reviewed and approved the final manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.