
Abstract
In cancer treatment, immunotherapy has great potential for improving the prognosis of patients with hematologic and solid malignancies. In this study, various bioinformatics tools and servers were used to design an antiangiogenic fusion protein. After comprehensive evaluation, an antiangiogenic fusion protein was designed using a soluble extracellular domain of human vascular endothelial growth factor receptor 1 (sVEGFR-1) and human interleukin-2 (IL-2) joined by a flexible linker. The final construct was composed of 875 amino acids. The secondary structure of the fusion protein, obtained by CFSSP, PSIPRED, and SOPMA tools, consisted of 14.17% helices, 29.71% extended strands, 4.69% beta turns and 51.43% random coils. Tertiary structure prediction by Raptor X showed that the fusion protein comprises 3 domains with 875 modeled amino acids, out of which 26 positions (2%) were considered disordered. The Ramachandran plot revealed 89.3%, 7.1%, and 3.6% amino acid residues in favored, allowed, and outlier regions, respectively. Physical features of the Molecular Dynamic (MD) simulated system such as root mean square deviation, root mean square fluctuation, solvent-on hand surface region, and radius of gyration identified the fusion construct as a stable and compact protein with few fluctuations in its overall structure. Docking of the fusion protein showed that interaction between sVEGFR-1/VEGFA and IL-2/IL-2R still exists. In silico analysis revealed that the fusion protein comprising IL-2 and sVEGFR-1 has stable structure and the selected linker can efficiently separate the two domains. These observations may be helpful in determining protein stability prior to protein expression.
Keywords
Introduction
The foremost cause of mortality around the world is cancer. Despite great advancements in the field of molecular oncology—improving both diagnosis and therapy—the mortality rate still remains high. 1 For the identification of cancer hallmarks, many approaches are currently being applied. Among biological cancer treatments, one of the features is to prevent the effect of tumor-promoting growth factors, hence inhibiting angiogenesis. Another aspect is to initiate an antitumor immune response by recruiting the immune effector cells around the tumor.2-4 However, significant advancements in the field of genetic engineering have led to the evolution of fusion proteins. All naturally occurring oncogenic fusion proteins are membrane-bound or cytoplasmic signaling molecules. No oncogenic fusion protein composed of extracellular ligands or cytokines is reported, so the fusion of extracellular ligands with signaling molecules, such as leukins, is attractive for pharmaceutical drug development. Research is in progress concerning the development of fusion proteins as a drug which may be administered as an exogenous compound acting upon certain target tissues. Different research groups have synthesized artificial fusion proteins as immunomodulators to forestall the development and progression of autoimmune diseases and cancer. 5 Gene fusion techniques are employed to produce recombinant proteins bearing the features of parental products. 6 These fusion proteins have diverse utilization in biomedicine, agriculture, pharmaceuticals, the food industry, and so on. Among fusion proteins, recombinant receptor decoys are constructed based on the fusion of the extracellular domain (ECD) of the receptor and Fc-part of the human IgG immunoglobulin. This fusion leads to improve the pharmacokinetic properties of the fusion protein, and thus is widely used in pre-clinical and clinical studies. However, the chimeric decoy receptor fusion protein is produced by fusing the two ECD entities or plasma membrane-associated receptors that may target their dedicated components individually (ie, ligands), inhibiting tumor-associated angiogenesis processes. 7 The study is related to the creation of a fusion protein borne of the marriage of two compounds; one is a cytokine named IL-2 and the second part comprises extracellular domain of VEGFR-1. Both proteins have an antiangiogenic effect, so it was proposed that this fusion can alter immune response and function with an enhanced antiangiogenic effect.
Shibuya and coworkers discovered VEGFR-1 as fms-like tyrosine kinase or Flt-1. 8 Flt-1 is a vascular endothelial growth factor receptor 1 (VEGFR-1). 9 Flt-1, now also known as VEGFR-1, was shown to be highly expressed in vascular endothelial cells. 10 The VEGFR-1 gene has two alternative polyadenylation sites: one is present in intron 13, and the other one after exon 30, which is the last exon of the gene. The alternative transcript encodes two isoforms: a soluble VEGFR-1 (sVEGFR-1) which lacks cytoplasmic kinase domains and without transmembrane domains, and the second isoform comprises a full-length transmembrane domain that shows weak kinase activity upon binding with VEGFA. 11 VEGFR-1 traps ligands through its ligand binding domain, therefore plays a negative role during angiogenesis. The primary function of VEGFR1 is to prevent the initiation of signaling cascades through the maintenance of less functional VEGFR2. In adults, VEGFR-1 is expressed not solely on the surface of endothelial cells but however conjointly on macrophages and promotes the function of macrophages, inflammatory diseases, cancer metastasis, and atherosclerosis via its kinase activity. VEGFR-1 as well as its soluble form is involved in a variety of human illnesses, making it very important target in the development of new strategies to suppress disease. 12
Cytokines are small glycoproteins which function by binding to the cell surface receptors and ultimately regulate the development, survival, and function of immune cells. Cytokines have been studied as therapeutic agents to check the response of the immune system to tumor cells. IL-2 is among the cytokines that exhibit pleiotropic effects on the immune system. After IL2 was discovered as “T cell growth factor” (TCGF) in 1976, it revolutionized immunology and immunotherapy. 13 IL2 was one of the first drug candidates approved by the FDA for cancer immunotherapy, for metastatic renal cell carcinoma in 1992 and for metastatic melanoma in 1998.14-17 IL-2 is 15.5 kDa, a four helical structure cytokine that is predominantly produced by antigen-simulated CD4+ T-cells as well as by CD8+ cells, natural killer (NK) cells, and activated dendritic cells (DC).18-20 The important functions of IL-2 are the maintenance of CD4+ regulatory T-cells, and differentiation of CD4+ T-cells. It can also promote CD8+ T-cell and NK cell cytotoxicity activity, and help in T-cell differentiation when exposed to antigen, thus enhancing the differentiation of naive CD4+ T-cell into two types: T helper-1 (Th1) and T helper-2 (Th2) cells. During this inhibition, differentiation of T helper-17 (Th17) takes place.21-23 The IL-2 receptor is composed of the three subunits: IL-2Rα (CD25), IL-2Rβ (CD122), and IL-2Rγ (CD132). The trimeric complex composed of αβγ subunits brings the affinity of the receptor to its highest level.24-28 IL-2 has displayed many examples of tumor regression, but it is not effective in improving patient survival rates due to dual-functional effects on T-cells and the adverse effects of high doses. As such, IL-2 monotherapy is insufficient for treatment of both metastatic renal cell carcinoma and metastatic melanoma. Researchers are now focusing on improving the efficacy of IL-2 therapy by instead using a combination of IL-2 therapy and other anticancer agents. 16 As soluble VEGFR-1 receptor can act as decoy receptor to scavenge ligands that are involved in angiogenesis without initiating an immune response, so we decided to fuse this s-VEGFR-1 with IL-2, both of these act as antiangiogenic agents, so a step toward developing fusion protein with enhanced antiangiogenic effect.
The 3D structure of proteins provides information regarding their folding pattern, stability, and interaction among domains. In designing the recombinant multi-domain proteins, the fusion proteins are more prone to misfolding or assembling in an incorrect 3D shape when compared with a single-domain protein, as different peptide domains interact with each other in different ways. 29 To overcome this limitation, in silico analyses are performed to generate the recombinant fusion proteins followed by the protein modeling and Molecular Dynamic (MD) simulations. In the current report, molecular modeling of an antagonistic angiogenic fusion protein—ie, soluble ECD of human VEGFR-1 (sVEGFR-1) and human IL-2—uses threading assembly refinement to predict the 3D structure and energy minimization. MD simulation is done to optimize the model and to observe its structural fluctuations which might be utilized for cancer therapy.
Materials and Methods
The current study involves the fusion of ECD of sVEGFR-1 (732 residues) and IL-2 (133 residues) by a flexible linker (10 residues) to build a fusion protein (875 residues), using structural modeling and then its characterization by MD simulation and molecular docking analyses.
Sequence retrieval and fusion protein modeling
The amino acid sequences of ECD of sVEGFR-1 and human IL-2 were retrieved from UniProtKB: VEGFR-1 id: P17948-2 (https://www.uniprot.org/uniprot/P17948) and Human IL-2 id: P60568 (https://www.uniprot.org/uniprot/P60568), respectively. The fusion protein consists of two chains, A and B. N-terminal A-chain consists of 732 residues and C-terminal B-chain consists of 133 residues. To build a fusion protein, the EC domain of VEGFR1 was linked with IL-2 using a hydrophobic and flexible (GGGGS)2 amino acid linker. Flexible linkers are used when there is requirement of movement at a certain degree or interaction between individual components of fusion protein. They are mainly composed of small non polar (eg, Gly) or polar (eg, Ser or Thr) amino acid residues. 30 The chimeric gene was designed for cloning and expression of the fusion protein in E. coli.
Primary structure analysis of fusion protein
ExPASy’s ProtParam server 31 was used to observe the physiochemical characters of fusion protein inclusive of theoretical isoelectric point (pI), molecular weight, molecular formula, total number of positive and negatively charged residues, instability index, 32 aliphatic index, 33 and grand average hydropathicity (GRAVY). 34 The instability index gives an estimate of a protein’s stability in vitro. The aliphatic index of a protein is regarded as a positive factor for the growth of thermostability of globular proteins, and specifically defined as the relative volume occupied by aliphatic side chains (alanine, valine, isoleucine, and leucine). The GRAVY score is calculated as the sum of the hydropathy values of all the amino acids, divided by the number of residues in the sequence.
Secondary structure prediction
The secondary structure of the ECD of sVEGFR-1 and fusion protein were determined using CFSSP (Chou and Fasman secondary structure prediction), 35 PSIPRED, 36 and SOPMA 37 servers. Moreover, the functional characteristics of the designed protein including secondary structure, the regions lacking regular shape, coiled-coil domain names, sections with low-complexity, transmembrane (TM) helices, solvent-on hand surface region (SASA), and the sites with disulfide bridges have been additionally assessed.
Three-dimensional (3D) model prediction
The RaptorX server was used for 3D structure prediction by homology modeling, and PDB ID: 5t89 crystal structure was used as a template. Protein molecules achieve maximum stability at their lowest energy state (through proper folding) to form a 3D structure. The RaptorX, 38 structure prediction server was used for automated prediction of protein secondary structures, template-based tertiary structure modeling, and probabilistic alignment sampling. Given a target sequence, the RaptorX server can detect even distantly related template sequences by using a novel algorithm utilizing probabilistic consistency and nonlinear, context-specific alignment potential. Raptor X assigns confidence scores by which quality of predicted 3D structure can be evaluated. P value indicates the quality of the predicted protein model. The smaller the p value, higher the quality of the predicted model.
Tertiary structure validation
UCSF Chimera alpha 1.14v was utilized to visualize and analyze the 3D predicted models, followed by mode assessment through the RAMPAGE tool. 39 It generates the Ramachandran plot which depicts and distributes the residues in favored, allowed, and outlier regions.
Molecular dynamics simulation
GROMACS program version 2021.2 was used for molecular dynamics of fusion protein.40,41 Optimized potential for liquid simulations (OPLS) force field was used and the structure was placed in a cubic unit cell with 1.0 nm from the box edge. Simple point charge (SPC) water model was used for solvation and then neutralization of system was done by Cl- ions that were substituted from solvent molecule. After that a maximum of 50 000 steps of energy minimization was performed of already predicted models by using a conjugate gradient algorithm followed by steepest descent minimization. The total energy should be constant throughout the simulation process, as it is the sum of kinetic and potential energy of the molecules. Kinetic energy should be constant or following a decreasing trend since the constant increasing of kinetic energy level reflects the general confusion of protein structure. Potential energy level should be increasing or constant to show the stability of structure.
After energy minimization, the system was equilibrated using the position restrained simulation under an NVT ensemble (constant Number of particles, Volume, and Temperature) for 100 picoseconds that stabilizes the temperature at 300 K with Berendsen thermostat followed by an NPT ensemble (constant Number of particles, Pressure, and Temperature) for 100 picoseconds to stabilize the pressure at 1.0 bar with Parrinello-Rahman pressure coupling factor.
At the end, unrestrained MD simulation was carried out for 100 nanoseconds with 2 fs per step with Berendsen thermostat of 300 K and the pressure at 1.0 bar with Parrinello-Rahman pressure coupling factor.
Comparative analysis of the structural deviation such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and solvent accessible surface area (SASA) were computed using GROMACS associated utility packages.
Protein-protein interaction (PPI)
After simulation, the molecular docking analysis between fusion protein and sVEGFR-1/VEGFA and IL-2/IL-2RA and IL-2RB was performed by using the ClusPro 2.0 tool for protein-protein docking. 42 The PPI has three computational steps: rigid body docking using the FFT (Fast Fourier Transform) correlation approach; RMSD-based clustering of structures generated to find the largest cluster that represents the likely models of the complex; and refinement of selected structures. The ClusPro 2.0 generates four types of output models using the scoring algorithms designated as balanced, electrostatic-favored, hydrophobic-favored, and van der Waals electrostatic. The first 10 relatively low-energy docking structures were selected and analyzed further for interaction analysis. UCSF Chimera alpha 1.1v Chimera software was used for visual representation, assessing the complex interactions, and measuring the distances among the interacting amino acid residues.
Results
Primary structure analysis
The primary structure of VEGFR-1 (p17948-2) is composed of 732 amino acid residues, encoded by means of 2196 nucleotides. Human IL-2 (Accession number: p60568) is composed of 133 amino acid residues, encoded by 399 nucleotides. The 732 amino acids fragment of VEGFR-1 (mature part) and 133 amino acid fragments of IL-2 were used to design the fusion protein construct. The length of the designed construct is 875 amino acids, which between 1 and 732 is VEGFR-1. Amino acids 733-743 (10 aa) are linker sequences, (GGGGS)2. Amino acids 744 to 875 (133 aa) are IL-2 (Figure 1A). Figure 1B shows a schematic view of the designed fusion protein. The primary structural features of the designed fusion protein determined with the help of ProtParam are summarized in Table 1. ProtParam is an application that lets in the computation of various physical and chemical parameters for a given protein. The calculated isoelectric point (pI) for VEGFR-1, IL-2, and fusion protein were calculated to be 9.15, 7.05, and 9.04, respectively, suggesting the presence of greater negatively charged residues. The value of the aliphatic index for VEGFR1, IL-2, and fusion protein were 84.29, 103.38, and 86.23, respectively, while that of the instability index were 48.62, 47.37, and 52.75. GRAVY (grand average hydropathy) values of VEGFR1, IL-2, and VEGFR1 (GGGGS)2 IL-2 were −0.348, −0.378, and −0.171, respectively.

(A) A Schematic view of the designed fusion protein, (B) A proposed model showing mode of action of the sVEGFR1-linker-IL2 fusion protein.
Amino acid composition.

Physiochemical properties of the fusion protein
The physiochemical properties of the fusion protein sequence were revealed using the ProtParam webserver. It was found that fusion protein is composed of 875 amino acid residues, the average molecular weight 98 kDa, and pI 9.04. The total number of negatively charged residues (Asp + Glu) was 85 and the total number of positively charged residues (Arg + Lys) 106. Percentage of different amino acid residues is shown in Table 1. The instability index of the fusion protein was computed by the ProtParam webserver (Table 2).
Parameters calculated by ExPSy’s ProtParam tool.

Abbreviations: AI, aliphatic index; EC, extinction coefficient at 280 nm; GRAVY, grand average hydropathy; II, instability index; Mw, molecular weight; −R, number of negative charged residues; +R, number of positive charged residues; TpI, theoretical Isoelectric point.
Prediction of secondary structure of fusion protein [sVEGFR-1-(GGGGS)2-IL-2]
The secondary structure of fusion protein [sVEGFR-1-(GGGGS)2-IL-2] predicted by CFSSP, 35 PSIPRED, 36 and SOPMA 37 tools was found to consist of 14.17% helices, 29.71% extended strands, 4.69% beta turns, and 51.43% random coils. The location of flexible linker (GGGGS)2 reside within amino acids 733-742 in the fusion protein (Figure 2).

Schematic illustration of secondary structure prediction of fusion protein [sVEGFR-1-(GGGGS)2-IL-2] by PSIPRED.
Prediction and validation of tertiary structure of fusion protein
The RaptorX server was used for 3D structure prediction (Figure 3) by homology modeling, using crystal structure PDB ID: 5t89X as a template. The fusion protein consisted of 3 domains, comprising 875 amino acids, out of which 26 (2%) positions were predicted as disordered. The best template was 5t89X with P value 2.35e-32, which was low and statistically significant. Overall uGDT (GDT) was 702(80). Solvent access values were 47% E, 34% M, and 18% B.

Predicted 3D structure of fusion protein [sVEGFR-1-(GGGGS)2-IL-2] showing α-helices, extended strands, β-turns and random coils, VEGFR1(green), IL2 (yellow), linker (black), VEGF (orange).
Analysis of simulation
After completing the simulation, energy (kinetic, potential & total energy), RMSD, RMSF, solvent accessible surface area (SASA), and Gyration radius were analyzed with output data.
Structural model refinement
Molecular dynamics simulation was used for structural refinements described in Materials and Methods. Simulation serves as a bridge between theory and experimentation. The theory was tested by simulation using computer-generated models that gave an idea about the possible strong interactions between molecules.42-45 Gromacs is an application of molecular dynamics simulation developed by Groningen University. Gromacs is specialized to perform MD simulation and energy minimization. The MD simulation output data were analyzed based on energy (kinetic, potential, & total energy), RMSD, RMSF, SASA, and Gyration radius.
Root mean square deviation
RMSD is the measure of the average distance between the backbone atoms of proteins. The structural refinement was carried out using molecular dynamics simulation over the equilibration course and exhibited RMSD plots for predicted models of fusion protein that flattened after 10-100 ns (Figure 4). RMSD plots indicated that there is very small deviation in the backbone of protein structure and is stable during simulation.

Root Mean Square Deviation plot of sVEGFR-1, IL-2 and fusion protein [sVEGFR-1-(GGGGS)2-IL-2], showing the deviations of Cα-atoms of proteins. The fluctuations of sVEGFR-1, IL-2 and fusion protein [sVEGFR-1-(GGGGS)2-IL-2] are highlighted in red, black and blue colors respectively.
Root mean square fluctuation
To analyze the mean atomistic motions of separate residues, root mean square fluctuation (RMSF) of Cα atoms in fusion protein was calculated using final trajectories. High RMSF values denote more flexibility (more conformational fluctuation), while low values show less fluctuations in the structure. As shown in Figure 5, the fluctuations in the fusion protein structure suggested that the orientation of the individual proteins in the fusion protein is quite stable, with less structural changes.
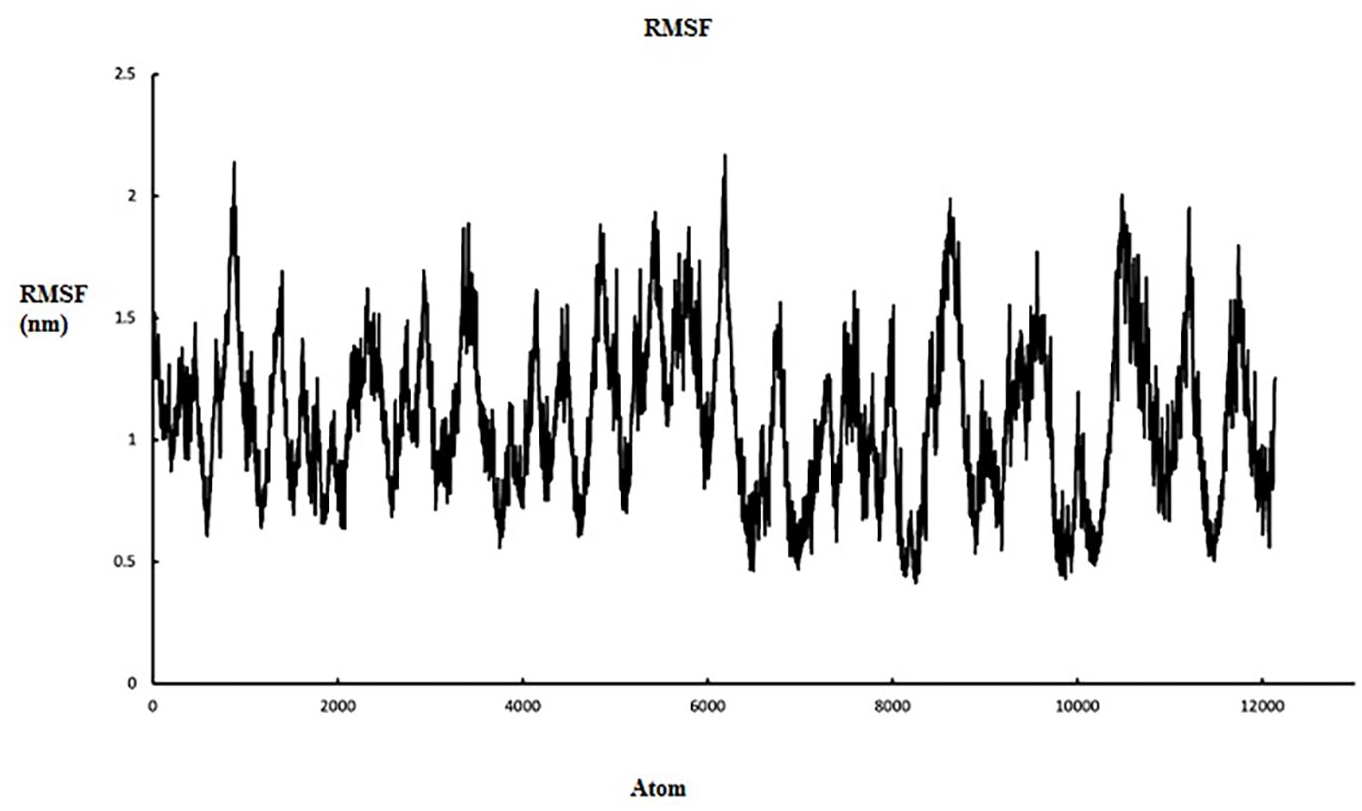
5 Root Mean Square fluctuation graph of sVEGFR-1, IL-2, and fusion protein [sVEGFR-1-(GGGGS)2-IL-2] to represent the residual fluctuations with time. The deviation lines of sVEGFR-1, IL-2 and fusion protein [sVEGFR-1-(GGGGS)2-IL-2] are highlighted in black, red and blue colors respectively.
Solvent accessible surface area
In addition to other interactions within the protein, very important intermolecular interaction is hydrophobic interaction. Hydrophobic interactions are present between non-polar amino acids and ensure the stability of proteins in solution by masking the non-polar amino acids present in the core. 46 UV fluorescence spectroscopy has been used to monitor hydration of hydrophobic core during protein unfolding and hydrophobic collapse during the start of protein folding.47,48 This spectroscopy technique exploits the intrinsic fluorescence property of proteins to provide sensitive indications of variation in the solvent accessibility of the hydrophobic core caused by changes in tertiary structure.49-52 During denaturation, unfolding of proteins inevitably causes the hydrophobic core to be exposed to the aqueous surrounding leading to the loss of hydrophobic interactions among non-polar amino acid clusters. Theoretically, changes in the accessibility of protein to solvent can be determined by computing solvent accessible surface area (SASA). During the course of the simulations conducted, the SASA of the fusion protein will naturally get larger as hydration of the hydrophobic core occurs during unfolding causing the interruption of hydrophobic interactions among non-polar residues. The plot of SASA (Figure 6) of the fusion protein showed decreasing trends indicating less exposure of the hydrophobic core to solvation as unfolding proceeds, making protein highly stable.

Solvent accessible surface area of sVEGFR-1, IL-2, and fusion protein [sVEGFR-1-(GGGGS)2-IL-2].
Gyration radius
Gyration radius is an indicative of the level of compaction in the structure, ie, how folded or unfolded is the protein. During 100 ns of simulation, this number was almost constant indicating the stability of the molecular structures. Figure 7 represents simulation analysis of fusion protein that validated the accuracy of our designed fusion protein structure.

Gyration radius graph of sVEGFR-1, IL-2, and fusion protein [sVEGFR-1-(GGGGS)2-IL-2].
Ramachandran plot assessment
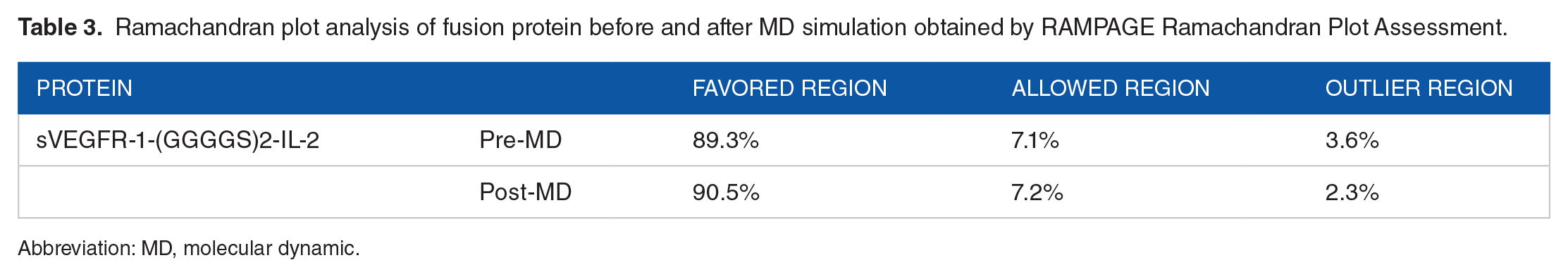
The 3D structure of fusion protein [sVEGFR-1-(GGGGS)2-IL-2] was validated by the Ramachandran plot before and after MD simulation, and structures were assessed by using RAMPAGE. The data showed the overall refinement of modeled structures as the number of amino acids in favored and allowed regions were improved, while residues in outlier region were reduced after a 100 ns simulation. The Ramachandran plot analysis obtained by RAMPAGE Ramachandran Plot Assessment is summarized in Table 3 and the plots are provided in Figure 8.
Ramachandran plot analysis of fusion protein before and after MD simulation obtained by RAMPAGE Ramachandran Plot Assessment.
Abbreviation: MD, molecular dynamic.
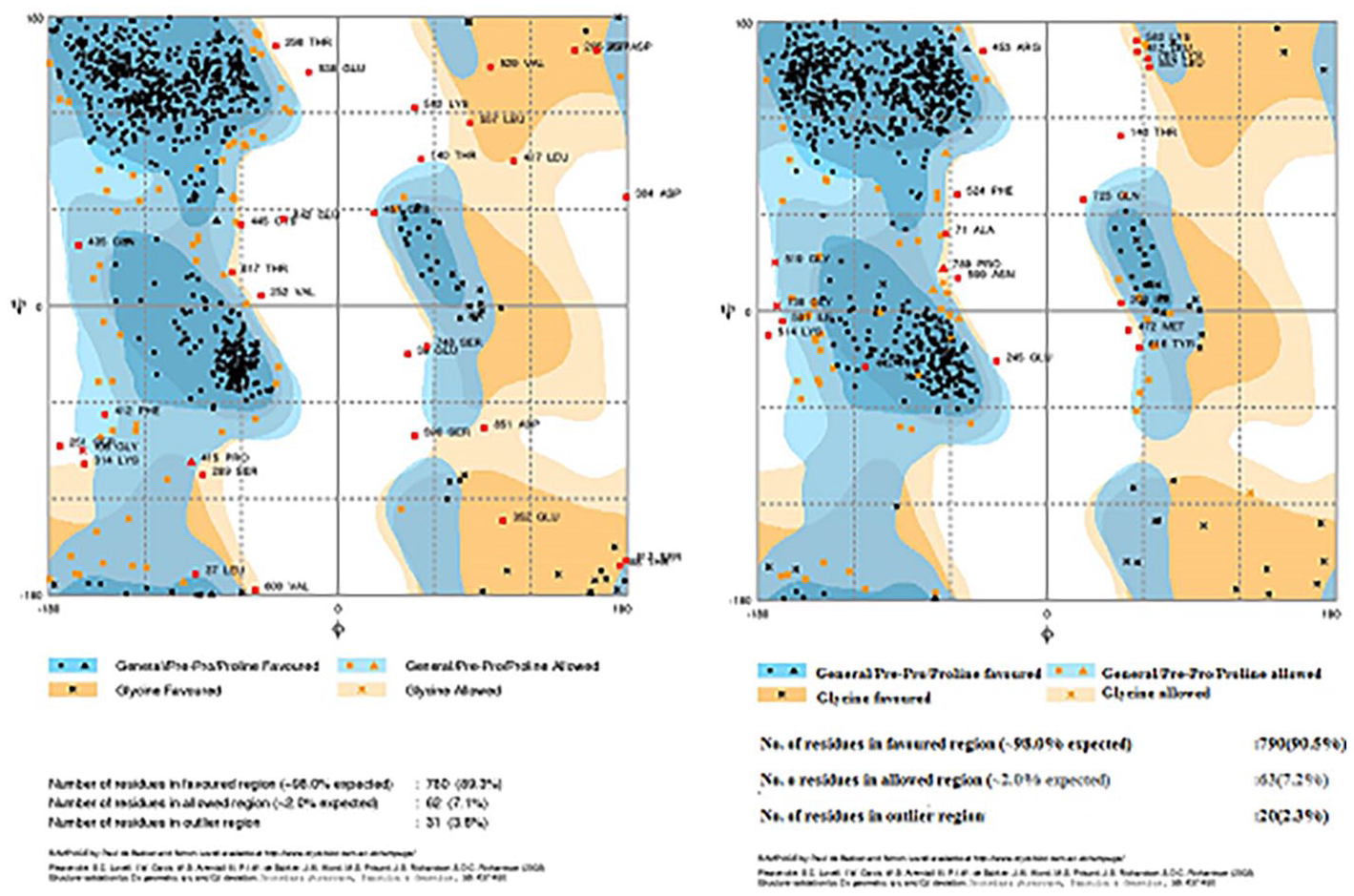
Ramachandran plot analysis of fusion protein [sVEGFR-1-(GGGGS)2-IL-2] before and after MD simulations. The general favored region and Pre-Pro favored region are indicated with dark blue color. The generally allowed region and Pre-Pro allowed region is shown in pale blue. The glycine favored and allowed regions are shown in dark and pale orange, respectively. The disallowed region is in white color.
Comparison of modeled structures before and after MD simulation
The 3D structure of fusion protein was also assessed after MD simulation for accuracy. By overlaying 3D structure of the fusion protein before and after MD simulation depicted that the predicted structure is stable, and the linker is able to separate the individual domains of the fusion protein. Structural comparison of predicted models of fusion protein before and after MD simulation is shown in Figure 8 and confirms the proper folding of fusion protein. The results of the Ramachandran plot suggested that overall, the quality of the predicted fusion protein model is satisfactory.
Protein-protein interaction of fusion protein [sVEGFR-1-[GGGGS)2-IL-2] with VEGFA, IL-2RA and IL-2RB
Docked complexes (ie, fusion protein with VEGFA and fusion protein with IL-2RA and IL-2RB) were analyzed by PDBSum 53 and then protein-protein interactions were interpreted by using visualization tools. A total of 21 hydrogen bonds and 4 salt bridges were found between IL-2RB and IL-2 (Figure 9). Five hydrogen bonds were observed between IL-2RA and IL-2, among them 3 amino acid residues were found to be involved in salt bridge formation (Figure 10). Moreover, the full docked complex of the fusion protein showed the presence of one salt bridge and 10 hydrogen bonds; of which, one of the hydrogen bonds was present between THR123 and GLN 37 (Figure 11). These results showed that even after the formation of the fusion protein, the interactions between IL-2RB/IL-2, IL-2RA/IL-2, and sVEGFR-1/VEGFA are still present. The detailed atomic interaction of IL-2RB/IL-2 (Table S1, S2, S3), IL-2RA/IL-2 (Table S4, S5, S6), and sVEGFR-1/VEGFA (Table S7, S8, S9) are mentioned in Supplementary data.

(A) Interacting residues of IL-2RB are highlighted in red, sVEGFR-1 in blue and IL-2 in dark green color. (B) Chain A is showing IL2RB and chain B represents IL2. (C) The focused image of interacting residues having salt bridges, disulphide bonds and hydrogen bonds between IL-2RB and IL-2.

(A) Interacting residues of IL-2RA are highlighted in red, sVEGFR-1 in blue and IL-2 in dark green color. (B) Chain A is showing IL2RA and chain B represents IL2. (C) The focused image of residues having salt bridges, disulphide bonds and hydrogen bonds between IL-2RA and IL-2.
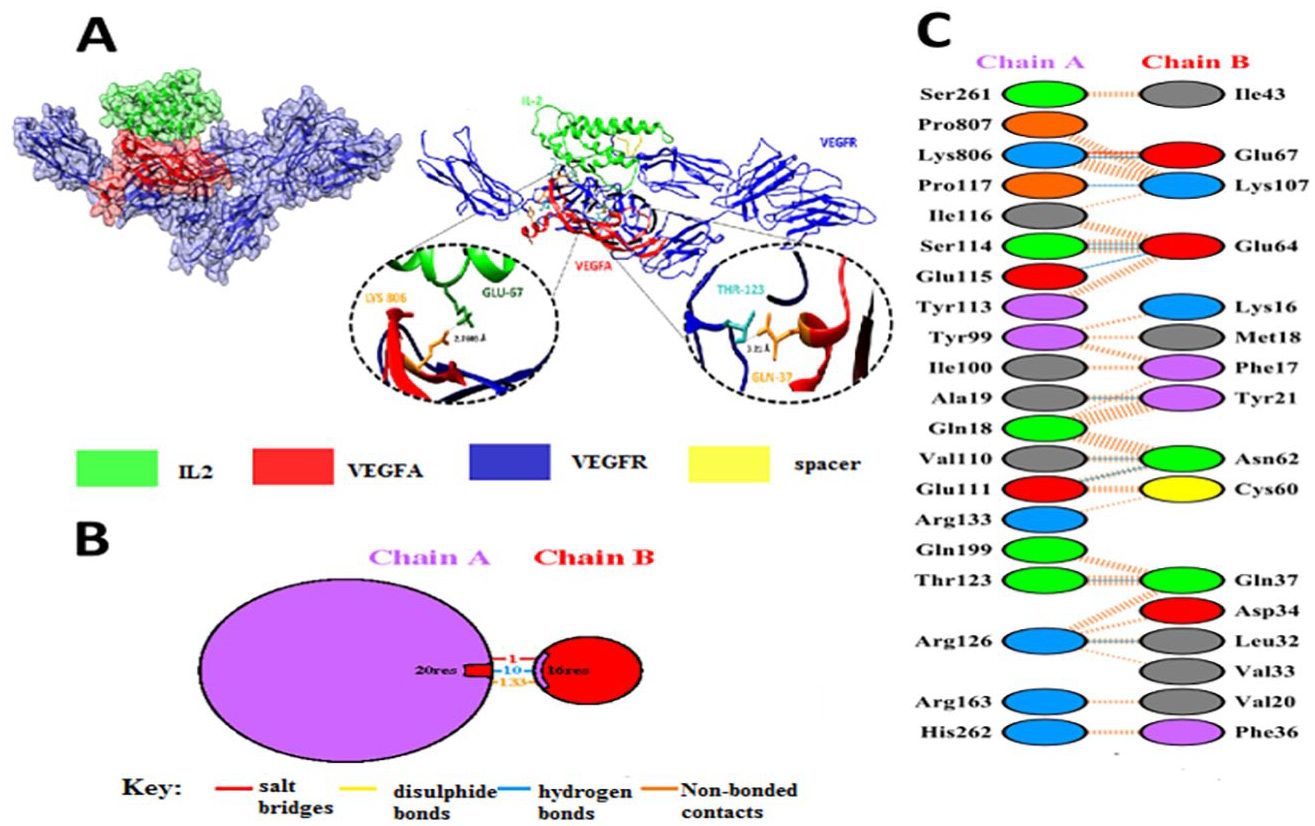
(A) Full docked complex of fusion protein [sVEGFR-1-(GGGGS)2-IL-2], IL-2 in dark green and VEGFR-1 in blue. (B) Chain A is showing sVEGFR-1 and chain B represents VEGFA. (C) The focused image of residues having salt bridges, disulphide bonds and hydrogen bonds between sVEGFR-1 and VEGFA.
Discussion
Cancer is the leading cause of death around the world. Immunotoxins are used in targeted therapies which target specific antigens or receptors on the surface of tumor cells. Targeted therapies where ligands are specifically targeted show more specificity to tumor cells as well as less toxic reaction.54-61 Recombinant DNA techniques have permitted fusions of genes in a simple way. In our work, in silico analysis of a chimeric fusion protein comprising sVEGFR1 and IL-2 is described.
Cytokines are the molecular messengers through which communication between different cells of the immune system is accomplished. They generate a synchronized, robust, and self-limited response to the targeted antigen. They act by efficiently propagating immune signals. Over the last two decades, scientific interest has piqued over the possibility of eradicating cancer by harnessing the immune system, exploring the cytokine characteristics and their signaling networks for cancer treatment. 62 The fusion of two or more cytokines with diverse biological activities may produce a unique fusokine with unheralded biopharmaceutical and therapeutic synergy properties that are not observed by individual moiety. 5 Keeping in view the antiangiogenic effect of both IL-2 and sVEGFR-1, it was hypothesized that the fusion of IL-2 and sVEGFR-1 could not only promote activation of IL-2 receptor-expressing cells, but also scavenge VEGFA and B ligands in these cell compartments.
Molecular modeling and simulation methods are used to predict conformation of a molecule at equilibrium, and how molecules change their configuration from one to another. 63 The 3D structure of sVEGFR-1 is not available in the protein database, so in silico analysis could be helpful to determine its 3D structure. Molecular modeling was performed to find the structure and properties of sVEGFR-1, and then fused with IL-2 by a flexible linker. Afterward, in silico analysis was carried out to confirm the proper folding of each domain in designed fusion protein. A linker fragment VEGFR-1-(GGGGS)2-IL-2 was designed to link both genes. The length of the designed construct was 875 amino acids, which between 1 and 732 is VEGFR1. Amino acids 733-743 (10 aa) are linker sequences, which is (GGGGS)2. Amino acids 744 to 875 (133 aa) are IL-2. Based on previous studies, N-terminal of VEGFR-1 64 and C-terminal of IL-2 65 seem more important in their biological function than the other end. Therefore, we designed the fusion protein in a way to make free the N-terminal of VEGFR-1 and C-terminal of IL-2. The primary structural features of the designed fusion protein were determined by ProtParam. The calculated isoelectric points (pI) for VEGFR1, IL-2, and VEGFR1 (GGGGS)2 IL-2 suggested the presence of more negatively charged residues. The value of the aliphatic index, instability index and GRAVY (Grand average hydropathy) values suggested a hydrophilicity pattern and better interaction with water.
The RaptorX server was used for 3D structure prediction by homology modeling. The fusion protein consisted of 3 domains, comprising 875 amino acids, out of which 26 (2%) positions were predicted as disordered. The best template was 5t89X with P value 2.35e-32, which was low and statistically significant. Solvent access values were 47% E, 34% M, and 18% B. The glycine-rich peptide linker in the fusion protein [(GGGGS)2] confers the flexibility and allows the two proteins (sVEGFR and IL-2) to move independent of one another by retaining their distinct 3D structure. 44 The linker fragment between two domains of the fusion protein can provide proper flexibility and separation, and the fusion protein after expression can provide proper accumulation in the periplasmic space of suitable host cell, because signal peptide is presented at the N-terminal of fusion protein which will permit it to cross the cytoplasmic membrane. Thus, the fusion protein will be secreted to the culture media and inclusion body formation would not occur. 66
The 3D structure of fusion protein [sVEGFR-1-(GGGGS)2-IL-2] was validated by the Ramachandran plot before and after MD simulation, and structures were assessed by using RAMPAGE. This showed the validation and accuracy of post-molecular dynamics (MD) modeled structures over pre-MD modeled structures. Considering all above model evaluations, it was inferred that the post-MD structures were of good quality and suitable for further analysis. MD simulations were carried out to predict the core factors responsible for the stability of fusion protein. The RMSD, RMSF, and SASA plots identified the fusion protein as stable structure. The radius of gyration plot confirms the fusion protein as a stable and compacted construct with very few fluctuations in its overall structure. The results of the Ramachandran plot suggested that, overall, the quality of the predicted fusion protein model is satisfactory. Docked complexes (ie, fusion protein with VEGFA and fusion protein with IL-2RA and IL-2RB) were analyzed, which showed that even after the formation of the fusion protein, the interactions between IL-2RB/IL-2, IL-2RA/IL-2, and sVEGFR-1/VEGFA are still present.
Conclusion
This study will be helpful for rapid analysis of the computationally designed fusion protein before initializing the wet lab experiments. It could be concluded that this procedure is fast, simple, and inexpensive, especially for the users that are new in this field. In silico analysis of the fusion protein [sVEGFR-1-(GGGGS)2-IL-2] structure revealed that the interaction remains between individual proteins after fusion. Flexible linker separates sVEGFR-1 and IL-2 domains effectively to maintain their proper folding and allows them to move independently of each other. The individual components of fusion protein act like bispecific ligands to drive unique downstream signaling events. The interaction that results from the fusion has shown great potential in the stability of the protein. Furthermore, the fusion protein may modulate immune response by overcoming maladaptive biological processes that underlie deadly diseases like cancer.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322211043297 – Supplemental material for A Computational Approach to Modeling an Antagonistic Angiogenic VEGFR1-IL2 Fusion Protein for Cancer Therapy
Supplemental material, sj-docx-1-bbi-10.1177_11779322211043297 for A Computational Approach to Modeling an Antagonistic Angiogenic VEGFR1-IL2 Fusion Protein for Cancer Therapy by Qurrat ul Ain Shafique, Hafiz Muzzammel Rehman, Tahreem Zaheer, Rana Adnan Tahir, Munir Ahmad Bhinder, Roquyya Gul and Mahjabeen Saleem in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Fond der Oesterreichischen Nationalbank OENB 15174.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Q.S. and R.G. designed the experiments. T.Z., H.M.R., and M.A.B. performed molecular modeling and docking analyses. R.A.T. and H.M.R. performed the molecular dynamic simulation analysis. Q.S. wrote the first draft. R.G. and M.S. conceived the idea and critically reviewed the manuscript. All the authors reviewed and approved the final manuscript.
Availability of Data and Material
The data used to support the findings of this study are included within the article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.