
Abstract
The incompleteness of partial human mitochondrial genome sequences makes it difficult to perform relevant comparisons among multiple resources. To deal with this issue, we propose a computational framework for deducing missing nucleotides in the human mitochondrial genome. We applied it to worldwide mitochondrial haplogroup lineages and assessed its performance. Our approach can deduce the missing nucleotides with a precision of 0.99 or higher in most human mitochondrial DNA lineages. Furthermore, although low-coverage mitochondrial genome sequences often lead to a blurred relationship in the multidimensional scaling analysis, our approach can correct this positional arrangement according to the corresponding mitochondrial DNA lineages. Therefore, our framework will provide a practical solution to compensate for the lack of genome coverage in partial and fragmented human mitochondrial genome sequences. In this study, we developed an open-source computer program, MitoIMP, implementing our imputation procedure. MitoIMP is freely available from https://github.com/omics-tools/mitoimp.
Introduction
The human mitochondrial genome has been used to study maternal phylogenetic relationships1-6 and disease-related mutations, 7 as well as for DNA identification, 8 and it is an important genomic region not only in molecular anthropology but also in the medical and forensic fields. High-throughput genotyping and parallel sequencing technologies have made it possible to perform human mitochondrial genome population analyses on a massive scale.9-11 These high-throughput technologies also allow mitochondrial genome analyses of ancient humans or archaic hominins, as well as modern humans.12-17 However, the quantity of endogenous DNA in archeological remains is extremely low, which makes it difficult to obtain complete mitochondrial genome sequences. As an experimental method to deal with this problem, several target enrichment approaches have been employed,18-21 but these problems are still not completely resolved in the cases of extremely degraded samples. The stability of endogenous DNA in postmortem resources is greatly affected by the environmental conditions, such as temperature, humidity, and pH. 22 A simulation study based on a statistical model suggested that the endogenous DNA in archeological samples is rapidly degraded in warm climates. 23 Actually, it is often difficult to obtain complete mitochondrial genome sequences from degraded samples excavated in areas with warm or hot climates, such as East or Southeast Asia.24,25
There are several practical approaches for analyzing incomplete data with missing values. One of these approaches is the listwise deletion approach, 26 which deletes categories, including missing values. However, this approach could limit comparisons with samples and discard some of the valuable data. Therefore, multiple imputation approaches have been proposed as methods to solve these problems.27,28 These approaches fill the missing values with approximate or estimated values, which allows samples to be analyzed without deleting categories including missing values. The imputation against the human mitochondrial genome facilitated the performance of mitochondrial haplogroup prediction, 29 improved the statistical power in a genome-wide association study, 10 and also minimized the impact of missing nucleotides in a population genetics analysis. 30 However, these previous studies did not sufficiently assess the effect of the imputation on the worldwide mitochondrial DNA lineages, and as far as we know, there is no available imputation tool designed for the human mitochondrial genome. In this article, we propose a computational approach for deducing the missing nucleotides in partial human mitochondrial genome sequences and also assessing the effects of the imputation on the worldwide human mitochondria DNA lineages. Our computational approach will provide a practical solution to compensate for the missing data in the low-coverage human mitochondrial genomes.
Materials and Methods
Simulated data set
To assess the impact of our approach on the global mitochondrial DNA lineages, we generated artificial low-coverage human mitochondrial genome sequences for representative mitochondrial macro-haplogroups (A, B, C, D, E, F, G, H, HV, I, J, K, L0, L1, L2, L3, L4, L5, L6, M, N, O, P, Q, R, R0, S, T, U, V, W, X, Y, and Z) in PhyloTree (Build 17; http://www.phylotree.org ). 31 Based on the mitochondrial genome sequences of these haplogroups, we simulated artificial next-generation sequencing (NGS) reads with ART (v. MountRainier-2016-06-05), 32 which is a simulation tool to generate synthetic NGS reads. In this study, we assumed paired-end reads based on the Illumina sequencer model in ART and mitochondrial genome coverage of ×10 to ×90. Next, these simulated short reads were aligned against the human mitochondrial reference sequence rCRS (revised Cambridge Reference Sequence), 33 using BWA (Burrows-Wheeler Aligner; v. 0.7.15). 34 We then obtained artificial low-coverage mitochondrial genome sequences from these aligned reads with MitoSuite (v. 1.0.9). 35 Finally, we applied our approach 500 times for each mitochondrial DNA lineage and investigated its precision. The precision of the imputation is computed as follows: TP/(TP + FP). True positive (TP) is the number of nucleotides imputed and validated. False positive (FP) is the number of nucleotides imputed but failed in validation.
Empirical Data Set
To assess the impact of our approach on the empirical sequencing data, we used several low-coverage human mitochondrial genome sequences from high-throughput sequencing data. These sequences were from Southeast Asian individuals dating from the Neolithic period through the Iron Age (4100-1700 years ago). 25 We downloaded these alignment reads according to the accession number PRJEB24939. Due to the poor DNA preservation in tropical conditions, most of the mitochondrial genomes derived from these ancient remains had incomplete partial sequences. Lipson et al 25 evaluated the ancient DNA authenticity and the exogenous contamination for each NGS library. To reduce the influence of postmortem misincorporations, 36 we clipped 2 bases from each end of the alignment reads, using BamUtil (v. 1.0.14). 37 Finally, we obtained the mitochondrial genome sequences from the clipped reads, using MitoSuite. In this study, the haplogroup assignments were performed according to the results of Lipson et al. 25
Computational Deducing Approach
Our deducing approach couples the allele-sharing distance used for low-coverage sequencing data 38 with the k-nearest neighbor (kNN), which is a simple and effective supervised classification algorithm. 39 First, our procedure computes the allele-sharing distance to determine the pairwise distance among individual mitochondrial genome sequences, which is given by the following expression:

where δij is the allele-sharing distance between individuals i and j, Sij is the number of polymorphic sites compared between individuals i and j, and

where M is the number of pairs in the individual mitochondrial genome sequences. Based on the distance matrix, the KNN sequences are selected, and then the missing positions are assigned to the most common alleles in the selected neighbors. To ensure the robustness of the imputed alleles, our framework also introduced the parameter f, which is the threshold frequency to determine the major alleles. In this study, we followed the parameter condition (f = 0.7, k = 5) used for kNN-based imputation procedures in Mizuno et al. 40 The condition is one of the most accurate combinations of parameter values in the previous research. Our approach also uses the reference population panel based on the complete mitochondrial genome sequences. Therefore, we obtained the worldwide human mitochondrial DNA sequences from PhyloTree Build 17 (http://www.phylotree.org). 31 These sequences were aligned using MAFFT (v. 7.407) 41 and oriented to the position of rCRS. Finally, we used 23 257 human mitochondrial DNA sequences as the default panel data set (ALL panel), including all present macro-haplogroups. The 1000 Genomes Project panel (phase 3) 42 is often used for imputation of genome-wide single-nucleotide polymorphisms (SNPs) in human population genetics, but the geographical region of the population and the mitochondrial genome lineage are limited. In this study, we designed the global panel data (ALL panel) with more than 4500 haplogroup lineages, covering known all macro-haplogroups. In addition, in our framework, there is no need to assign mitochondrial haplogroups of sequences in advance. Our approach does not force the design of panel data to be composed of the same population or maternal lineages and will be able to prevent false estimates from panels with biased population structure. To investigate whether the panel design can rescue lineage-specific mutations, we also used the worldwide macro-haplogroup panels consisting of the same macro-haplogroup lineages as the input mitochondrial genome.
We also developed an open-source computer program, MitoIMP, implementing our imputation procedure (Figure 1). MitoIMP is freely available from https://github.com/omics-tools/mitoimp. Our computer program supports the standard FASTA as the input and output format. In our computational framework, the genomic position of a mitochondrial genome sequence is oriented to that of the human mitochondrial genome reference sequence rCRS with MAFFT. 41 Finally, MitoIMP outputs an imputed mitochondrial genome sequence in the FASTA format and provides a summary table for the imputation procedure.

Flowchart of the imputation procedure.
Multidimensional Scaling Analysis
To assess the impact of our approach on an empirical low-coverage mitochondrial genome, we performed a multidimensional scaling (MDS) analysis using the mitochondrial genome sequences from 18 Southeast Asian ancient remains. The MDS analysis revealed a 2-dimensional relationship among the samples, based on the genetic distance of the mitochondrial genome sequences. The distances were determined based on the allele-sharing distance. 38 The MDS analysis was performed before and after our deducing approach. Pairwise allele-sharing distances between mitochondrial genomes were calculated using MitoIMP, and the eigenvector of the distance matrix was obtained by the “cmdscale” command, which is implemented in R. 43 The scatter plots were drawn using the ggplot2 package 44 in R.
Results
Application to simulated low-coverage sequencing data
To investigate the effectiveness of our approach to worldwide mitochondrial DNA lineages, we assessed the precision of the imputation for each maternal lineage. Our simulation results showed that missing bases can be estimated with a precision of 0.99 or higher in most of the macro-haplogroup lineages (Figure 2). We also found that the imputation precision for several macro-haplogroup L lineages was more affected by the loss of genome coverage than the other macro-haplogroups (Figure 2). Our approach showed decreased precision with the loss of genome coverage, but the FPs were only a few bases even where the mitochondrial genome lacked half of the genome coverage (Supplemental Figure S1). We also examined the region-by-region effects of our approach on the mitochondrial genome.

The assessment of imputation procedures for mitochondrial haplogroup lineages.
Although the imputation is uniformly performed on the entire mitochondrial genome, the effect of this procedure has not been evaluated so far for each region. Therefore, we also examined the precision of the imputation for each region of the mitochondrial genome and found that the effect of this procedure varied by region on the human mitochondrial genome (Figure 3). Although several nucleotide positions of the D-loop showed lower precision, our approach imputed the missing nucleotides with >0.9 high precision throughout most regions of the human mitochondrial genome.

The assessment of imputation procedures across the human mitochondrial genome.
Application to Empirical Low-Coverage Sequencing Data
We used partial mitochondrial genome sequences from 18 ancient South Asian individuals for the empirical deducing approaches. These partial mitochondrial genome sequences showed different genome coverages with 5831 to 16 558 nucleotides, which are equivalent to 35.2% to 99.9% of the covered region of the human mitochondrial reference sequence rCRS. 33 Especially, VN39 was not found in 10 731 positions, corresponding to about a 65% loss of the whole mitochondrial genome. To exclude intentional filling by geographical region–specific haplogroups, we used the worldwide haplogroup sequences (ALL panel) for deducing the missing positions (parameters: k = 5, f = 0.7). As a result, more than 99% of the missing positions were filled after the process (Table 1).
Empirical mitochondrial genome sequences used in this study.

The mtGenome coverage shows the percentages of mapped positions in the mitochondrial genome. Missing nucleotides represent the number of missing bases in the mitochondrial genome.
To investigate the relative relationships among individuals before and after the imputation, we performed an MDS analysis. The results of the MDS analysis without/with missing data showed different positional arrangements (Figure 4A). Among the partial mitochondrial genome sequences with missing data, there is no correspondence between the arrangement and their maternal lineages. The calculated distance matrix among the partial mitochondrial genome sequences did not correspond to the assigned haplogroups (Figure 4B). Our results also revealed that such partial mitochondrial genome sequences have a significant correlation between the calculated distance and the loss of coverage (Pearson correlation r2 = .548, P = 2.384e−13; Supplemental Figure S2). On the other hand, the mitochondrial genome sequences imputed by our approach presented at least 2 clusters, belonging to macro-haplogroups B and M7 (Figure 4A and B). The individuals belonging to these 2 clusters showed different positional relationships before the filling procedure, but the imputed mitochondrial genome sequences seem to be plausibly arranged among the assigned macro-haplogroups. This result indicates that our approach can correct the unclear 2-dimensional arrangements among partial mitochondrial genome sequences.
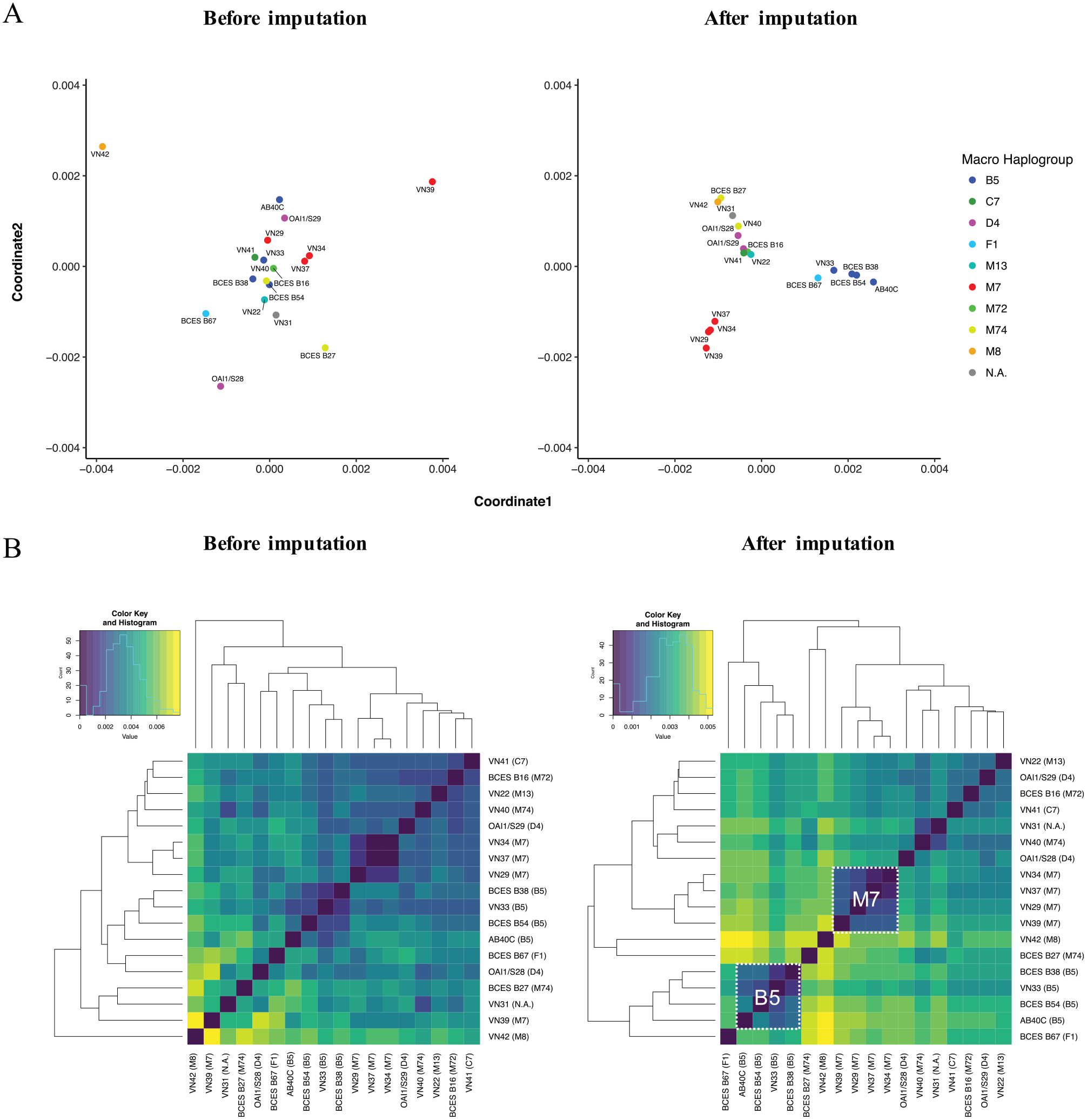
The relative relationships among individuals before and after the imputation. (A) The left and right figures show the results of the MDS analysis before and after the imputation procedure, respectively. The color scheme is according to macro-haplogroup lineages—B5 (blue), C7 (green), D4 (pink), F1 (light blue), M13 (cyan), M7 (red), M72 (light green), M74 (yellow), M8 (orange), N.A. (dark gray). (B) These figures are heat maps, based on the allele-sharing distance matrix among the empirical human mitochondrial genome sequences. Color keys of the distance values are shown on the upper left of each heatmap. The left figure shows the heatmap based on the allele-sharing distance before the imputation. The right figure shows the heatmap based on the allele-sharing distance after the imputation. Clusters of macro-haplogroups B5 and M7 are outlined by the white dashed lines.
Discussion
The effect of the loss of genome coverage
The missing data of mitochondrial genome sequences may be observed in empirical low-coverage sequencing data or genome-wide SNP array data. For example, ancient samples and museum samples have only trace amounts of endogenous DNA, and it is difficult to obtain complete mitochondrial genome sequences from such degraded samples. Although the incompleteness of the mitochondrial genome sequence may influence the calculation of the allele-sharing distances among individual sequences, our approach appears to be able to complement the deletion with the correct base in most of the haplogroup lineages. However, the effect of the imputation approach on the missing data in the human mitochondrial genome sequence may vary among the regions. The human mitochondrial genome consists of 37 genes and a control region (D-loop). These 37 genes comprise 13 subunits of the respiratory chain complex genes, 22 transfer RNA genes, and 2 ribosomal RNA genes. 45 Therefore, we should consider the effect of our approach across the entire region of the human mitochondrial genome sequence. In this study, we found several positions (nucleotide positions: 152, 310, and 16 519) with a precision less than 0.9. These positions are located on the D-loop regions, which are known to have a higher mutation rate than that of the gene-coding regions in the human mitochondrial genome. 46 As private alleles are accumulated in the D-loop, it is not preferable to use many candidate sequences in the KNN algorithm. Therefore, we may decrease erroneous estimations in D-loop by reducing the number of the parameter k. Although the effect of low coverage was observed at only a few nucleotide positions, most of the missing positions were correctly filled across the human mitochondrial genome sequence. This result indicates that our approach using partially obtained sequences helps to infer a nearly complete mitochondrial genome sequence. Our method will be a useful approach to complement the missing parts of low-coverage mitochondrial genome sequences in which unidentified sites occur randomly.
Understanding the Maternal Lineage for Accurate Imputation
To accurately fill the missing nucleotides in partial mitochondrial genome sequences, it is important to assess the impact of the imputation on each maternal lineage. Although the design of the population panel may lead to a bias of the filled nucleotides, a panel design with appropriate maternal lineages will improve the imputation performance. Actually, a previous study using 1500-year-old highly degraded samples showed that a closely related population panel (haplogroup A panel) can fill more missing sites, as compared with population panels including other maternal lineages. 40 However, this previous study assessed the impact of imputation on the macro-haplogroup A lineages, but that of the worldwide maternal lineages has not been verified. In this study, we used the population panels for the worldwide mitochondrial genome lineages and also tested the effect of these panels on the imputation performance. Our simulation result showed that the population panels consisting of closely related maternal lineages could improve the precision of the imputed nucleotides (Figure 2). Although the precision slightly varied due to the haplogroup lineages, our approach filled the missing sequences of the low-coverage mitochondrial genome with nucleotides with a precision of 0.99 or higher.
The imputation performance may also reflect the differences in the phylogenetic diversity of the haplogroup lineages. The most recent common ancestor of the human mitochondrial genome is dated back to approximately 150 000 to 250 000 years ago,2,3,47 and the phylogenetic relationships involving modern human mitochondrial genomes can be largely divided into maternal lineages in sub-Saharan Africa (African haplogroup L) and non-Africa, with the exception of the “back-to-Africa” lineages. 48 Especially, it is well known that the human mitochondrial DNA lineages among Africans are more than twice as diverse as those of non-Africans. 49 Therefore, although the precision of imputation might decrease in several haplogroup L lineages, our approach will be especially effective for most of the non-African mitochondrial DNA lineages. Depending on the maternal lineages of samples, setting the parameters may be useful to reduce biased alleles’ assignments caused by population structure. For example, population-specific alleles can be reduced by increasing the parameter k and the parameter f.
Impact of Deducing Approach for Poorly Preserved Fossil Remains
The full-length sequence of the human mitochondrial genome can facilitate the estimation of population dynamics,47,50,51 and new findings surrounding the diversity of the human mitochondrial genome have been revealed by comparisons with the diversity of mitochondrial genomes of archaic hominins, such as Neanderthals. 52 However, extremely degraded archeological remains often contain less than 1% endogenous DNA, which makes it difficult to obtain the full-length mitochondrial genome. Environments with high temperatures and humidity rapidly fragment the endogenous DNA in postmortem samples; therefore, ancient genome research involving warm and humid environments, such as East or Southeast Asia, has been hindered. As approaches to examine such degraded ancient remains, experimental methods such as DNA extraction protocols specialized for ancient bones53,54 and target enrichment methods19,55,56 are often applied. However, these experimental methods are generally expensive, and when applied to highly degraded fossil remains, the complete mitochondrial genome sequence would not be successfully obtained. Therefore, short regions, such as hypervariable regions (HVRs), have often been used in previous studies on ancient DNA from warm and humid geographical regions.57-59 However, the mitochondrial genome coverages obtained from ancient remains vary according to their DNA preservation. Actually, some empirical sequencing data from prehistoric Southeast Asian samples have large defects in the D-loop, including the HVRs. HV39 showed an especially low-coverage mitochondrial genome sequence, with more than half of the coverage missing. In addition, our simulation results suggest that the effect of the missing data on the control regions including HVRs is greater than that in other regions of the mitochondrial genome. Partial and incomplete mitochondrial DNA sequences might not be considered and neglected in detailed discussions. Therefore, we may also have limited insight into the analysis of low-coverage mitochondrial genome sequences including missing data. The filling of missing alleles for ancient genomes might be a challenging approach because ancient human genome resources are limited. However, our results show that our application can robustly impute most of the missing nucleotides in worldwide human mitochondrial DNA lineages (Figure 1 and Supplemental Figure S1). In this study, we have shown that the missing nucleotides can have a considerable impact on genetic distance calculations (Supplemental Figure S2). Our approach would be preferable to minimize the impact of missing nucleotides on the downstream analysis such as the MDS analysis.
In this study, we propose a computational deducing approach designed for such incomplete mitochondrial genome sequences. Recent improvements in NGS and target enrichment technologies have increased the opportunity to obtain randomly fragmented various partial sequences beyond specific regions of the human mitochondrial genome. Our approach imputes the missing regions using such partial sequences of the mitochondrial genome, without relying on the use of limited sequences or SNPs. Thus, this approach will work better for shotgun or target enrichment sequencing, as compared with genome-wide SNP arrays, amplicon sequencing, and polymerase chain reaction amplification. We also applied our approach to empirical low-coverage mitochondrial genome sequences. In the MDS analysis, our approach achieved the correct positional relationships by filling in the missing nucleotides. In particular, the individuals belonging to the macro-haplogroup lineages B5 and M7 appeared to be more closely clustered after the imputation. This result suggests that most of the missing regions were complemented by phylogenetically informative nucleotides. Our approach can be applied to low-coverage mitochondrial genome sequences from these empirical poorly preserved fossil remains and will prompt the use of partial mitochondrial DNA sequences that previously would have been discarded.
Supplemental Material
supplementary_files_xyz225878e63c6ee – Supplemental material for MitoIMP: A Computational Framework for Imputation of Missing Data in Low-Coverage Human Mitochondrial Genome
Supplemental material, supplementary_files_xyz225878e63c6ee for MitoIMP: A Computational Framework for Imputation of Missing Data in Low-Coverage Human Mitochondrial Genome by Koji Ishiya, Fuzuki Mizuno, Li Wang and Shintaroh Ueda in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
Discussions with our lab members greatly contributed to improving this study. Especially, we appreciate Dr Jun Gojobori and Dr Masahiko Kumagai for their expert advice.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by a Grant-in-Aid for JSPS Fellows 15J08490 (to K.I.), JSPS KAKENHI Grant Number 19K16246 (to K.I.), JSPS KAKENHI Grant Number 17K07588 (to F.M.), Zhejiang Provincial Natural Science Foundation of China Grant Number LZ13H020001 (to L.W.), and JSPS KAKENHI Grant Number 25291104 (to S.U.).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
KI contributed to the design and analysis of the research, to implementation of the source code and to the writing of the manuscript. FM, LW and SU provided critical feedback and helped shape the research, analysis and manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.