
Abstract
Plasmodium falciparum adenylosuccinate lyase (PfADSL) is an important enzyme in purine metabolism. Although several benzimidazole derivatives have been commercially developed into drugs, the template design as inhibitor against PfADSL has not been fully explored. This study aims to model the 3-dimensional (3D) structure of PfADSL, design and predict in silico absorption, distribution, metabolism, excretion and toxicity (ADMET) of 8 substituted benzo[d]imidazol-1-yl)methyl)benzimidamide compounds as well as predict the potential interaction modes and binding affinities of the designed ligands with the modelled PfADSL. PfADSL 3D structure was modelled using SWISS-MODEL, whereas the compounds were designed using ChemDraw Professional. ADMET predictions were done using OSIRIS Property Explorer and Swiss ADME, whereas molecular docking was done with AutoDock Tools. All designed compounds exhibited good in silico ADMET properties, hence can be considered safe for drug development. Binding energies ranged from −6.85 to −8.75 kcal/mol. Thus, they could be further synthesised and developed into active commercial antimalarial drugs.
Keywords
Introduction
Malaria is one of the most challenging infectious diseases to eradicate, especially in Sub-Saharan Africa. 1 Plasmodium falciparum remains the most prevalent malaria parasite in the world accounting for 216 million estimated cases in 2016. 2 The drug resistance of malaria parasite has led to the need and search for new chemical scaffolds that have novel modes of action and can act through new protein targets.3,4 One of such protein targets in P falciparum is the adenylosuccinate lyase (ADSL), which is an important enzyme in purine metabolism. 5 The de novo purine biosynthetic pathway that gives rise to the formation of adenosine monophosphate (AMP), catalysed by ADSL, is absent in P falciparum, making it a potential drug target for antimalarial studies.6,7 Cassera et al 7 reported that 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and its analogues can serve as potential inhibitors for ADSL of P falciparum, hence novel putative antiparasitic agents. Benzimidazole derivatives (substituted benzo[d]imidazol-1-yl)methyl)benzimidamides) were considered as potential analogues for AICAR due to similarities in chemical structure (Figure 1), and could be evaluated for their antimalarial propensity. Benzimidazole derivatives have been widely used in recent years due to their wide range of pharmacological activities including antimalarial, 8 antileishmanial, 9 analgesics, 10 anticancer, 11 antitumour, 12 antimicrobial, 13 anti-inflammatory, 14 antihepatitis C virus, 15 antihelmintic, 16 antibacterial 17 and antitrypanosomal 18 activities. Although several benzimidazole derivatives have been synthesised and developed into commercially available drugs, little is known about the design of the template as an inhibitor against P falciparum ADSL (PfADSL).
Structures of AICAR and benzimidazole showing the regions of similarity.
Over the years, different approaches have been used to improve how antimalarial agents are designed, put through clinical trials and eventually released as commercially available drugs. 19 One of such approaches is structure-based drug design (SBDD), which relies on the knowledge of the 3-dimensional (3D) structure of the protein target to design a suitable ligand that can function as its potential inhibitor. 20 In a situation where the experimental 3D structure of the protein is not available, homology model can be built from its amino acid sequence. 21 Molecular docking is an important technique in SBDD, which can be applied in facilitating and speeding up the development of antimalarial agents or drugs that can be active against the deadly malaria parasite. 22 Molecular docking has helped scientists to virtually screen a library of ligands (or compounds) against a target protein and predict the binding conformations and affinities of the ligands to the target. 19 The aim of this study is to model the 3D structure of PfADSL, design and predict the in silico absorption, distribution, metabolism, excretion and toxicity (ADMET) of some substituted benzo[d]imidazol-1-yl)methyl)benzimidamide compounds as well as predict the potential interaction modes and binding affinities of the designed ligands with the modelled PfADSL.
Materials and Methods
Homology modelling of PfADSL and the target-template sequence alignment
The experimental crystal structure of PfADSL is not available in the Protein Data Bank (PDB); 23 hence, its 3D structure was modelled. The protein ID of the target (P falciparum adenylosuccinate lyase 3D7 strain) was retrieved from UniProt Knowledgebase (UniProtKB) 24 with the accession number Q7KWJ4. Afterwards, the protein ID was submitted to SWISS-MODEL 25 web server to develop a model with sufficient query sequence coverage and sequence identity. The most reliable 3D structure was selected based on the Global Model Quality Estimation (GMQE) 26 and Qualitative Model Energy Analysis (QMEAN) 27 values. The GMQE values are usually between 0 and 1, and the higher the number, the higher the reliability of the predicted structure, while for QMEAN, a value below 4.0 shows reliability. 28 The similarity identity between the amino acid sequences of the homology model of PfADSL and the template structure used for the homology model were confirmed using Clustal Omega version 1.2.1. 29
Structure validation of modelled protein
The SWISS-MODEL web server automatically calculates the QMEAN scoring function for the estimation of the local and the global model quality based on the geometry, the interactions and the solvent potential of the protein model. It also provides the z-score ranging from 0 to 1, which are compared with the expected value for any structure. PROCHECK was used to check for the quality of the modelled 3D structure of PfADSL generated via SWISS-MODEL. For this structure validation, the .pdb file format of the modelled PfADSL was uploaded on the PDBsum web server 30 of European Bioinformatics Institute. The .pdb file format of the modelled PfADSL was uploaded on the server to obtain both the Ramachandran plot and the Ramachandran plot statistics. While the Ramachandran plot is used in accessing the quality of a modelled protein or an experimental structure, the Ramachandran plot statistics provides information on the total number of amino acid residues found in the favourable, allowed and disallowed regions. 31 Also, Verify3D 32 was used to validate the structure of the modelled protein, determine how compatible a 3D structure is to its own amino acids and compare the result with that of good-known structures.
Alignment of the PfADSL model and the template structure
The alignment of the PfADSL model and template structure was carried out using PyMOL molecular viewer 33 to show how closely related the carbon atoms are. This is derived from the root mean square deviation (RMSD) between the positioning of the carbon atoms of both the template and the model that is obtained from the alignment. The lower the RMSD (w.r.t 0), the more closely related the structures are.
Ligand modelling
AICAR analogues are good inhibitors of PfADSL
7
and similar to benzimidazole, as shown in Figure 1. Therefore, the benzimidazole derivatives were built as ligands to function as potential inhibitors of PfADSL, which is the target protein. The 2-dimensional (2D) structures of the substituted benzo[d]imidazol-1-yl)methyl)benzimidamide compounds,

Schematic representation of the compounds
In silico drug-likeness and toxicity predictions
Drug-likeness is a prediction that determines whether a particular pharmacological agent has properties consistent with being an orally active drug. 36 This prediction is based on an already established concept by Lipinski et al, 37 called Lipinski rule of five. The rule predicts that there is likely to be poor absorption or permeation when a compound possesses more than 5H-bond donors, 10H-bond acceptors, molecular weight greater than 500 and the calculated LogP (CLogP) greater than 5. 37 The selection of compounds as drug candidates is also determined by a parameter called drug score. 38 The higher the drug score value, the higher the chance of the compound being considered as a drug candidate. 38 The in silico drug-likeness and toxicity predictions of the designed ligands were carried out using OSIRIS Property Explorer 39 and Swiss ADME predictor.40,41 OSIRIS Property Explorer programme estimates the mutagenic, tumorigenic, irritant and reproductive risks, and also provides information on the compound’s hydrophilicity (LogP), solubility (LogS), molecular weight, drug-likeness and drug score. 42 Meanwhile, SwissADME predictor provides information on the numbers of hydrogen donors, hydrogen acceptors and rotatable bonds, total polar surface area and the synthetic accessibility of the compounds. The ligands were also subjected to Lipinski et al, 37 Muegge et al, 43 Ghose et al, 44 Egan et al 45 and Veber et al 46 screenings using SwissADME predictor. The analyses of the compounds were compared with that of chloroquine, and only compounds without violation of any of the screenings were used for the molecular docking analysis.
Protein preparation
The homology modelled 3D structure of the target protein, PfADSL, was downloaded from SWISS-MODEL in its .pdb format. The modelled protein structure was defined as receptor while the complexed ligands were removed using Chimera software. 47 Furthermore, the protein was prepared by the computation of Gasteiger charges, with the addition of polar hydrogens and merging of the nonpolar hydrogens using AutoDockTools 1.5.6. 48
Prediction of active sites in the modelled protein
The Computed Atlas of Surface Topography of proteins (CASTp) 3.0 49 was used to predict the active sites that were present in the modelled protein structure. CASTp is an online server that is applied in the identification and measurement of voids on 3D protein structures. 50 The modelled 3D protein was submitted on the server, and the necessary amino acids for binding interactions were predicted. 50
Molecular docking analysis
It has been reported that ADSL enzymes, which were used for the docking analyses, are biologically active as homotetramers.51,52 The molecular docking studies were carried out using AutoDockTools, which is a free graphic user interface (GUI) for the AutoDock4.2 programme. 53 The grid box was constructed using 58, 58, and 40, pointing in x, y, and z directions, respectively, with a grid point spacing of 0.508 Å. The centre grid box is of 14.527 Å, 56.689 Å and −5.122 Å around Arg 17A, Tyr 18A, Asn 312A, His 173C, Asn 90D, Asp 92D, Gln 250D, Arg 338D, Ser 343D and Arg 347D. These amino acids were selected based on the CASTp result and the alignment of the modelled 3D structure to the template structure. In addition, the docking analysis was executed using Lamarckian Genetic Algorithm 4.2, and the macromolecule was kept rigid throughout the docking simulation. The number of genetic algorithm runs was set at 10, and the other docking parameters were left at default values. Ten different conformations were generated for each ligand scored using AutoDock 4.2 scoring functions and were ranked according to their binding energies. AutoDockTools, PyMOL and LigPlot 54 were used for the post-docking analyses.
Results and Discussion
Homology modelling of PfADSL and the target-template sequence alignment
A 3D structure of PfADSL was built using SWISS-MODEL with GMQE of 0.80 and QMEAN of −1.46. Also, Plasmodium vivax ADSL Pv003765 with AMP bound (PDB ID: 2QGA; resolution: 2.01 Å) 55 was identified to have the closest template to PfADSL with a similarity identity of 63.91% and sequence similarity of 0.50. The GMQE value of 0.80 and QMEAN score of −1.46 indicate that the modelled structure is reliable and has a good quality.26,28
The multiple sequence alignment of the amino acid sequences 56 of the PfADSL (UniProtKB ID: Q7KWJ4) and P vivax ADSL with AMP bound (PDB ID: 2QGA) is shown in Figure 2. A percentage identity matrix of 63.36% was obtained, which confirms the similarity identity of 63.91% obtained from the homology modelling.

Alignment of the amino acid sequences of Plasmodium falciparum ADSL and the crystal structure of 2QGA.
Structure validation of modelled protein
The plot of the predicted local similarity to target against the residue number of the predicted 3D structure of the modelled protein was graphically represented (Figure 3A). The value of most of the residues was close to 1, indicating that the local quality estimate of the residues of the predicted model is good. The residues with values lower than 0.6 were considered to be of low quality. The modelled protein structure also lies within the range of other protein structures in PDB, which confirms its reliability (Figure 3B).
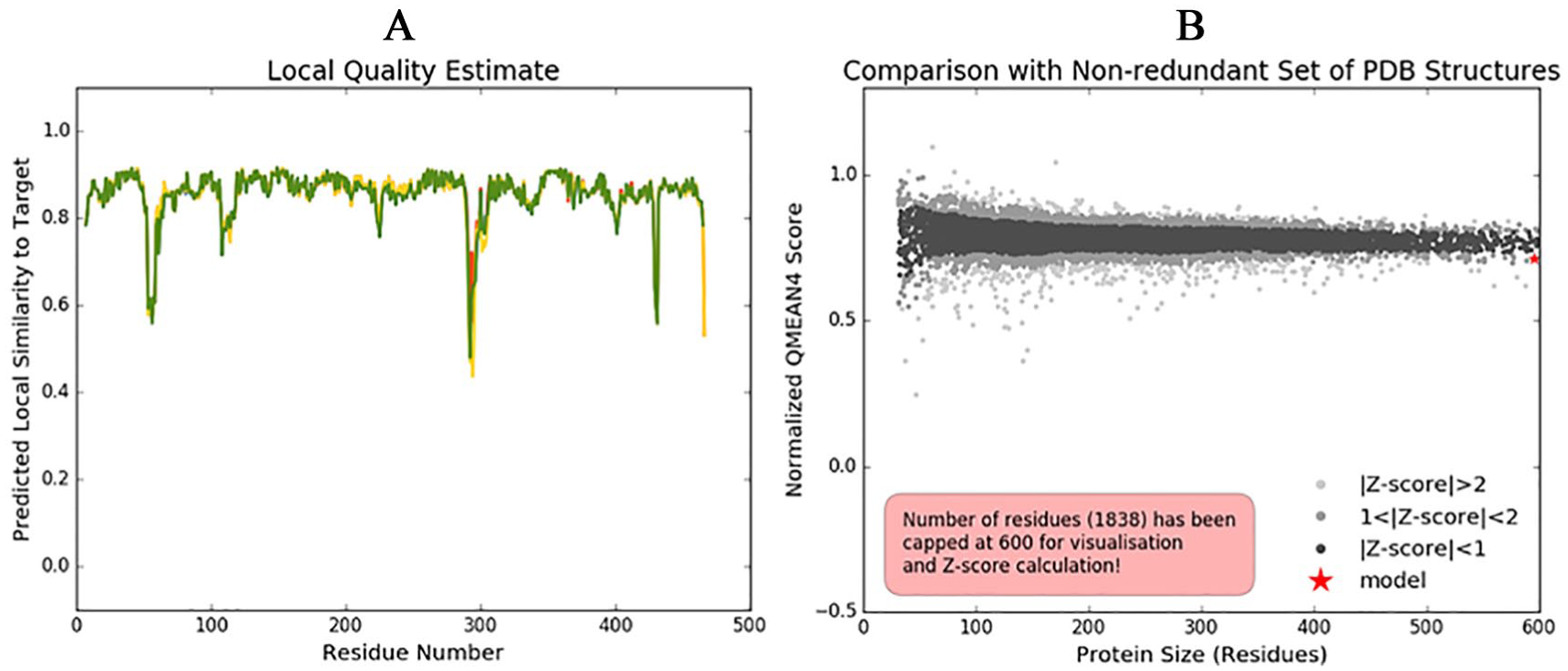
Structure validation of modelled PfADSL: (A) Local quality estimate of the residues of the predicted PfADSL model; (B) comparison of the predicted PfADSL structure with nonredundant set of PDB structures.
Both the Ramachandran plot (Figure 4A) and the Ramachandran plot statistics (Figure 4B) were obtained from PDBsum web server. The Ramachandran plot statistics implied that the modelled 3D structure of PfADSL has 91.8% of its residues in the most favoured regions, 7.4% of its residues in additional allowed regions, 0.8% of its residues in the generously allowed regions and 0.0% of its residues in disallowed regions of the Ramachandran plot. This also validates that the modelled 3D structure is a good quality model. Also, the Verify3D plot of the modelled protein (Figure 4C) was obtained for the structure validation and it showed as PASS. The 3D environment profile shows that 85.64% of the residues have averaged 3D-1D score ⩾ 0.2, which suggests the validity of the modelled protein.

Structure validation using (A) Ramachandran plot; (B) Ramachandran plot statistics of the homology modelled PfADSL; and (C) Verify3D.
Alignment of the PfADSL model and template (2GQA) structure
A RMSD value of 0.105 Å was obtained from the alignment computed using PyMOL molecular viewer, indicating that the structures were closely related (Figure 5). The template structure is represented by the blue helices, whereas the protein model is represented by the green helices. The alignment showed that the chain B and chain C of the dimer template structure (2QGA) corresponded to the chain C and chain D of the tetramer structure of the protein model. Meanwhile, it was observed from the molecular viewer that the binding of the AMP to the amino acid residues of 2QGA at His 168B, Asn 85C, Asp 87C, Gln 245C, Ser 338C, Arg 333C and Arg 342D also corresponded with the binding of the AMP with the amino acid residues of the modelled template at His 173C, Asn 90D, Asp 92D, Gln 250D, Ser 343D, Arg 338D and Arg 347D.

Alignment of the PfADSL model and the 2GQA template structure of Plasmodium vivax ADSL.
In silico results of risks and drug-likeness of ligands
a. OSIRIS property explorer result. With the exception of compound
Physicochemical properties and toxicity risks of compounds

b. SwissADME prediction
The numbers of hydrogen bond acceptors (NHA) and hydrogen bond donors (NHD) in compounds
ADME prediction of compounds

Abbreviations: ADME, absorption, distribution, metabolism, excretion; NHA, no. of hydrogen bond acceptors; NHD, no. of hydrogen bond donors; NRB, no. of rotatable bonds; TPSA, total polar surface area.
Active site identification
From the active site prediction, a pocket was identified with an area (SA) of 2919.055 and a volume (SA) of 2797.556 (Figure 6). A total of 166 amino acid residues were predicted to be the active sites for the modelled protein. However, the following were chosen as the more favourable sites for the docking analyses due to the similarities observed from the alignment of the modelled structure to the template structure: Arg 17A, Tyr 18A, Asn 312A, His 173C, Asn 90D, Asp 92D, Gln 250D, Arg 338D, Ser 343D, Arg 347D.

The surface of the binding pocket of the modelled protein as computed using CASTp 3.0.
Molecular docking results
The obtained binding energies and hydrogen bonds of compounds
Energy-based interactions and hydrogen bonds for benzimidazole derivatives

Abbreviations: AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; AMP, adenosine monophosphate.

Molecular docking interactions between AICAR and the binding sites of PfADSL: (A) 2D model of the interactions between AICAR and PfADSL; (B) 3D model of the interactions between AICAR and the binding sites of PfADSL.

Molecular docking interactions between
Furthermore, all the designed compounds exhibited dock score values between −6.85 and −8.75 kcal/mol, having lower binding energies than that of the complexed ligand (AMP) that had a binding energy of −5.10 kcal/mol. Also, the binding energies of the compounds were lower than AICAR (–5.49 kcal/mol), which has been reported to be a potential inhibitor of PfADSL.
7
The lowest autodock score and the best interactions were used to ascertain the compound with the best conformation.
2
The best dock score among the designed benzimidazole derivatives was −8.75 kcal/mol for compound
Conclusion
PfADSL is a potential drug target that can be considered in the design of antimalarial compounds to combat the malaria menace. This study gives an insight into the design and prediction of potential interaction modes and binding affinities of 8 substituted benzo[d]imidazol-1-yl)methyl)benzimidamide compounds with homology modelled PfADSL. (E)-4-((2-styryl-1H-benzo[d]imidazol-1-yl)methyl)benzimidamide,
Footnotes
Acknowledgements
The authors acknowledge Covenant University for the infrastructural support of this work and also thank Jide Ayodele of Covenant University Bioinformatics Research (CUBRe) for assisting with logistics.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Fogarty National Institutes of Health Common Fund (Grant No: 1U2RTW010679) and Alexander von Humboldt (AvH) Senior Georg Forster for EA.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
BB and EA determined the choice of the protein target. EA, OOA, and GOO put up the general concepts and design of the study. GOO carried out the implementation of these concepts. GOO, OOA, YUA and EA carried out the analysis of the work. GOO wrote the paper. GOO, OOA, YUA and EA revised the manuscript.