
Abstract
The tetratricopeptide repeat (TPR) of proteins consists of a 34-amino acid, alpha-helical motif that comprises a pattern of small and large hydrophobic residues, leading to a recognizable signature sequence. Structural and functional studies have documented that tandem TPRs form a superhelix that interacts with client molecules through strategically placed amino acids. Interestingly, most of the known TPRs are flanked by alpha-helices that lack the TPR signature but often appear as a continuation of the TPR superhelix. The exact role and specificity of these TPR-accompanying non-TPR helices have remained a mystery. Here, starting with TPR proteins of known structure, bioinformatic analyses were conducted on these helices, which revealed that they are diverse in sequence, lacking a clear consensus. However, they display significant atomic contacts with the nearest TPR helix and, to some extent, with the next TPR helix over. The majority of these contacts do not use the signature residues of the TPR helix but rather involve hydrophobic side chains on the facing sides. Thus, compared with the TPR helices, these companion helices are generic in nature, and seem to serve as relatively passive gatekeepers, leaving the terminal TPR helices to encode the signature residues that interact with cognate clients.
Introduction
The tetratricopeptide repeat (TPR) is a degenerate sequence of 34 amino acids that occurs in different proteins in tandem repeats. Search for TPR repeats has led to their identification in a large number of proteins in a wide variety of species. 1 These studies also showed that the number of TPRs in a protein can range from 1 to more than a dozen, although 3 repeats (TPR1-TPR2-TPR3), designated as 3TPR, were the most abundant.2-4 Each unit TPR motif consists of 2 helices, generally designated A-helix and B-helix, each 13 to 14 residues long (Figure 1), although these lengths are somewhat variable.

An idealized TPR and its companion helices (TPR-companion helices or TCHs). This is a schematic diagram, largely based on previous publications.3,5 As shown, the TPR is eponymously made of 34 residues and contains 2 helices (A, B). The accompanying upstream and downstream helices are designated as uTCH and dTCH, respectively. The TCH residues are independently numbered, ending with N, because of their variable distances from the TPR and varied lengths. The most conserved residues in the TPR-signature motif are indicated underneath (Trp4, Gly8, Tyr11, Ala20, Pro32).
Being an all-helical domain, the TPR domain bears close structural similarity with other alpha-helical domains. In fact, in the Structural Classification of Proteins (SCOP), it is a member of the “TPR-like” superfamily, which in turn belongs to the “alpha-alpha superhelix” fold in the “all alpha protein” class. Detailed analysis of naturally occurring TPR domains, TPR-containing full-length proteins, and consensus synthetic TPR peptides (abbreviated CTPR) has revealed the general architecture of TPR as an all-helical right-handed superstructure2,5,6 in which the A- and B-helices are packed at an angle of ∼24°.
However, TPR is distinguished from other helix-turn-helix proteins in several aspects, 3 of which are most relevant here: a characteristic consensus sequence, a superhelical structure, and the so-called “solvation helix.” Early studies, 7 and more recent large-scale survey of a few hundred 3TPR domains,3,4 revealed an approximate but recognizable consensus, in which small and large hydrophobic residues occur in similar positions in all TPRs, even though no 1 position was invariant (Figure 1). Nonetheless, several residues, particularly W4, Y11, A20, and P32, are relatively conserved,3,4 and therefore serve as TPR signature. As the TPRs generally occur in multiple tandem repeats in natural proteins, ranging in number from 3 to 40 (eg, in GenBank Q6PGP7) or possibly more, the overall TPR domain curves into a superhelix, in which the inner concave surface is largely involved in interaction with other proteins, and the specificity of each TPR domain is determined by a combination of the surface geometry and contact with specific residues.3,4 In-depth analysis of the CTPR, containing 1, 2, or 3 TPR motifs, has suggested specific structural and roles of the some of the signature residues, 3 likely conserved in all TPRs.
Finally, as briefly noted earlier, essentially all 3TPRs have a companion non-TPR helix at the end of the run of TPRs, the sequence of which, unlike those of the TPR helices, do not have conserved signature residues.3,6 Although an exact role of the 3TPR downstream helix is yet to be resolved, its presence in the synthetic TPR peptide facilitated the solubility of the peptide, and thus, it was named “solvating helix” or “solvation helix.”3,5 These studies implicated that the solvation helix interacts with the TPR in some manner, likely with the terminal TPR, which is the closest in distance; however, due to the absence of any structure-function analysis of the solvation helix in the context of a full-length natural TPR protein, its biological role remained a mystery. In the current study, bioinformatic analysis focused on the interactions between this non-TPR helix and the terminal TPR to determine the extent and distance of such interactions, to find out whether there is a pattern of the mutually interacting residues. As this study was undertaken, it was noticed that many TPR-domain proteins contained additional helices that were TPR-upstream. To start without any bias of their function and difference of function, both types of helices have been referred to as “TPR-companion helix” (TCH) and also as uTCH and dTCH to indicate upstream and downstream of the TPR domain, respectively.
Methods
Collection and display of TPR structures
Experimentally determined 3-dimensional (3D) structures of TPR proteins, such as from X-ray crystallography or solution Nuclear Magnetic Resonance (NMR), were obtained by querying the RCSB Protein Database (PDB; https://www.rcsb.org/) with “TPR.” From 341 structures, the following were eliminated: redundant structures (ie, multiple structures of essentially the same protein, deposited by different groups); “TPR oligomerization domains” (eg, 5TO7, 5TO6, 5TO7, 5TVB); consensus, idealized, or “redesigned” synthetic TPR (eg, 2FO7, 5FZR, 2HYZ); TPR in complex with other proteins (eg, 4APO, 4AIF, 4YVQ) or small molecule ligands (many examples), because they might affect TPR-TCH interactions; domain-swapped TPRs (4YMR); entries (eg, 3HA4, 4BSX) that did not confirm the repeat by in silico repeat motif search (such as by TPRpred, https://toolkit.tuebingen.mpg.de/#/), or contained Pentatricopeptide Repeat (PPR); (eg, 2MHK), a similar but distinct tricopeptide-family repeat (consisting of 35 amino acids; also predicted by TPRpred); half-TPRs (eg, 5IC8); incomplete sequence or regions with no assigned secondary structure (perhaps due to low-quality diffraction or disordered regions); and entries of mutant variants of the wild type, but with identical TPR sequence, and therefore effectively redundant sequences. The selected structures were downloaded as PDB files and displayed and analyzed using PyMol, 8 as stated in the corresponding figure legends (eg, Figures 2 and 3).
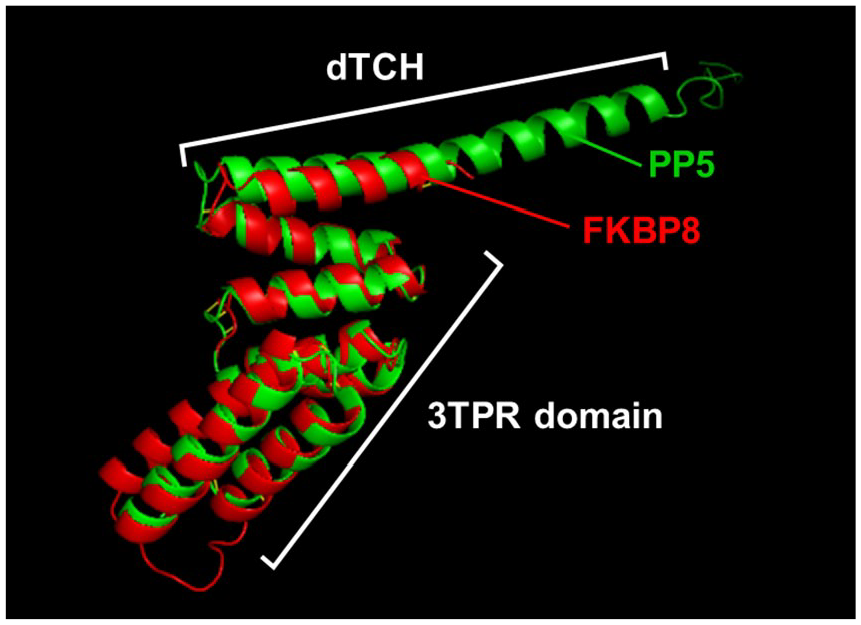
Conservation of structure in TPRs and TCHs. Two representative TPR domains with their cognate downstream TCHs, belonging to PP5 (1A17, in green) 2 and FKBP8 (5MGX, in red), 9 were superimposed by PyMol, which shows strong structural similarity when the extra-long helical segment of the PP5 TCH is excluded. TCH indicates TPR-companion helices; TPR, tetratricopeptide repeat.
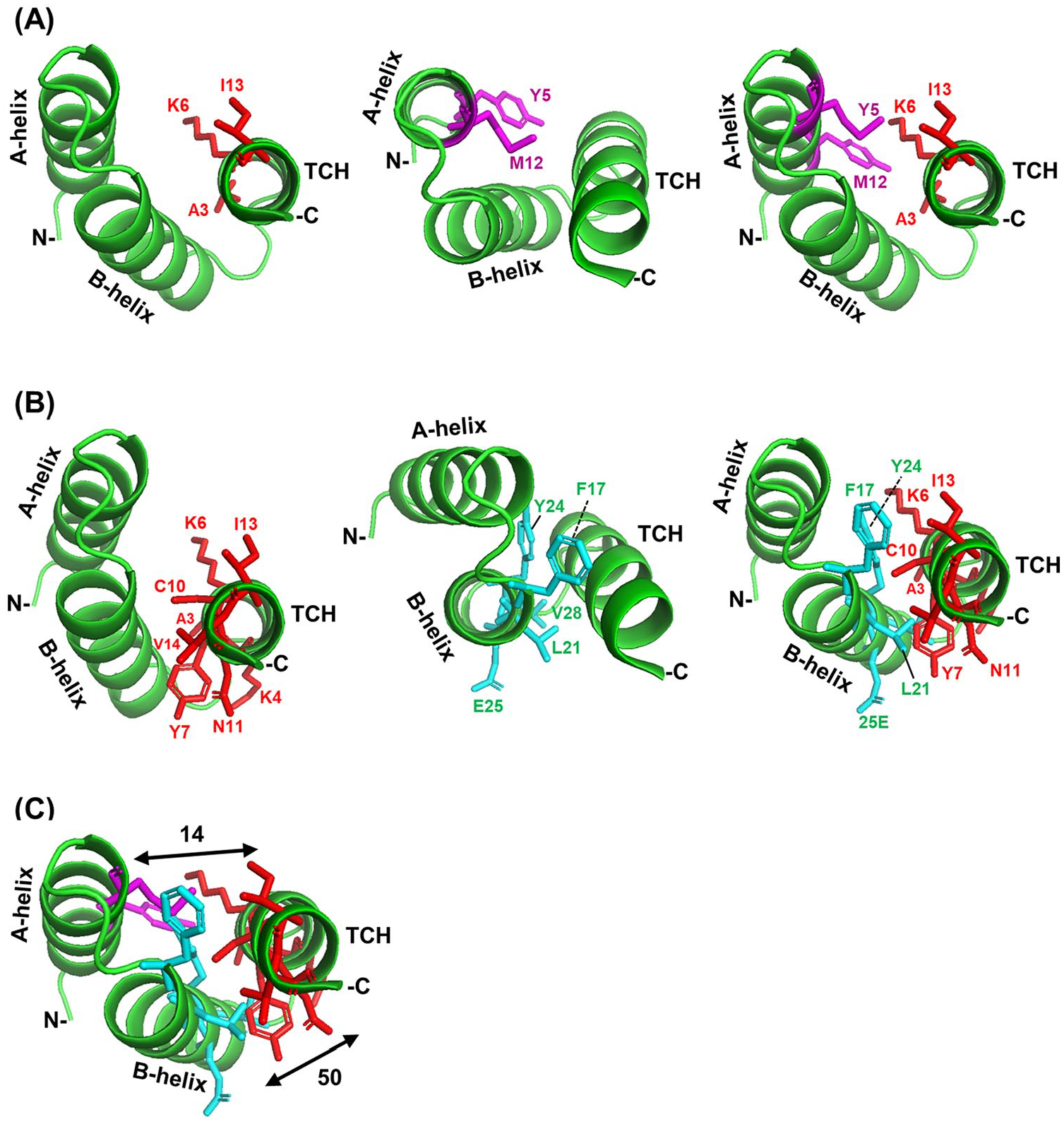
Interactions of TCH and TPR helices in PP5. The A- and B-helices of the terminal TPR (TPR3) of human PP5 (PDB 1A17) 2 along with the downstream TCH are presented as green ribbons and the interacting amino acid side chains as sticks of distinctive color (TCH, red; A-helix, magenta; B-helix, cyan). The amino (N) and carboxy (C) terminal directions are so marked. The amino acids are indicated with position numbers and single letter codes. (A) Interactions between TCH and A-helix. The interacting side chains from left to right are TCH side chains, A-helix side chains, and side chains of both helices together. (B) Interactions between TCH and B-helix. The interacting side chains from left to right are TCH side chains, B-helix side chains, and side chains of both helices together. (C) Combination of panels A and B, to depict pairwise interactions among the 3 helices together. The numbers indicate the total number of contacts between 2 interacting helices. Note that TCH makes many more contacts with the B-helix (50 contacts), which is closer, than with A-helix (14 contacts). In each panel, the same PyMol structure is rotated at different angles to make it easier to visualize the helices, their orientations, and the interactive amino acid side chains. TCH indicates TPR-companion helices; TPR, tetratricopeptide repeat.
Analysis of atomic contacts
The PDB structures were analyzed for specific TCH contacts in the MRC Protein Contacts Atlas web server (https://www.mrc-lmb.cam.ac.uk/rajini/index.html) 10 geared toward the depiction of non-covalent bonds in proteins, ideally suited for this study. The TCH, visually detected in the PyMol display, was confirmed in the sequence and the structure display. Helices that showed interaction with the TCH were then identified in the “chord” plot, and amino acids and the number of contacts were obtained from the interaction matrix. They were also visually confirmed in the interactive 3D structure. All residues showing >1 contacts were collected.
In a complementary approach, the sequences were also examined in terms of the free energy of interaction at the Amino Acid Interaction Web Server (http://bioinfo.uochb.cas.cz/INTAA/). 9 INTAA utilizes a molecular mechanical force field and generates an Interaction Energy Matrix (IEM), listing contacts made by each amino acid and their interaction energies. The IEM method is based on the hypothesis that amino acids with the most stabilizing interactions contribute to the folding enthalpy, and thus, it determines the key residues that are important for stability and correct protein folding. This site also offers interactive 3D structure with interaction energy colors. As the various non-covalent contact bonds have wide range of thermodynamic energy, for the sake of priority, a baseline of −4 (negative 4) was chosen. In other words, the major contacts with energy values of magnitude >4 were collected (see Table 2) and then visually confirmed in the interactive 3D structure from the Protein Contacts Atlas web server mentioned above. Although −4 was an arbitrarily chosen number, its choice as baseline did not affect the conclusions, as most of the interactions were much stronger (Table 2).
Multiple sequence alignment
Several multiple sequence alignment (MSA) suites, namely, Clustal Omega, Kalign, Multiple Alignment using Fast Fourier Transform (MAFFT), MUSCLE, and T-Coffee, all publicly available at EMBL-EBI (https://www.ebi.ac.uk/Tools/msa/), were used to find a common signature among the 141 TCH sequences from proteins of known structures. Although they use a variety of algorithms such as Hidden Markov Model (HMM) profile-profile (eg, Clustal Omega) and Fast-Fourier Transforms (eg, MAFFT), and optimized for different sequence lengths, all are suitable for protein alignment and can detect and display amino acid identities as well as conservative replacements.
Results
The search for a consensus sequence in the TPR-companion helices
To find any similarity in the TCH amino acid sequences, TPR proteins whose structures have been experimentally determined were first collected (Table 1), so that the location and sequence of the helices were rigorously defined. In a comprehensive previous study of the 3TPR sequences, a synthetic 15-mer sequence (AEAKQNKGNAKQKQG) was used, which apparently worked equally well to promote solvation of 1, 2, or 3 identical copies of the consensus TPR. 3 As mentioned earlier, the 3TPR domains as a class have garnered the most attention in consensus studies, mainly due to their abundance in nature and the relative uniformity of their superhelical structure.3,4 For this reason, the 3TPRs were placed in 1 group (Panel A in Table 1), and TPR numbers smaller or greater than 3 in a separate group (Panel B in Table 1).
TCH in experimentally determined three-dimensional structures.

Abbreviations: PDB, Protein Database; TCH, TPR-companion helix; TPR, tetratricopeptide repeat.
This table presents only those TPR proteins for which the structure has been determined experimentally, such as by X-ray crystallography or NMR. The PDB numbers of the structures are shown. The 3 TPR groups (A-C) are described. In TPRs with repeat numbers other than 3 (B), the number of repeats is indicated in parentheses. Each sequence includes the full-length TPR (underlined) as well as the full upstream and/or downstream TCH (uTCH, dTCH) (highlighted in gray), as appropriate. A20 is a major signature amino acid in TPRs and, where found, is also highlighted in gray to serve as landmark. Proline residues between uTCH and the TPR are highlighted in green (closer to uTCH) or pink (closer to A-helix). Red colors mark mutually contacting residues of the TCH and the A- and B-helices, which are detailed later under the “Helical location of the TPR-TCH interacting residues” section.
Due to the unknown role of the uTCH and their rarer occurrence, only 2 examples are listed and were not studied in further detail.
Even a cursory look at the TCH sequences made it apparent that irrespective of the number of TPRs in the protein, a consensus TCH signature may not be found. First, the primary structures as well as locations of the TCHs are obviously very diverse. Second, their lengths were also dissimilar, which presented uncertainty in performing a multiple alignment. Also note that they were placed at varying distances from the B-helix, although this may not pose a major hurdle in the interaction between the 2 helices because these interhelical regions were unstructured and hence likely flexible. Nevertheless, attempts were made to perform multiple alignment of the TCH amino acid sequences by established programs, as described in the “Methods” section, all of which failed to generate any consensus (data not shown).
It could be argued that the failure to find a TCH consensus sequence was due to the relatively small number of TCH structures that have been experimentally determined. The search was then extended to a larger cohort by including 115 bioinformatically predicted structures, using Solvent AccessBiLitiEs (SABLE) (Figure S1). To make the study fully inclusive, a sundry mix of naturally occurring repeat numbers, ranging from 1 to 40, was collected. As shown (Figure S1), the A- and B-helices of the last TPR motif could be easily recognized by their TPR-signature residues. In contrast, the predicted TCHs, downstream of the B-helix, once again presented a disparate cohort of no clear homology. As with the experimentally determined TCHs, their lengths were also diverse, ranging from 6 to >33. In a few cases, the prediction algorithm failed to detect any helical structure in the region (eg, in P09913, Q13099, Q1L5Z9, B7Z5B3, and F4MH46) (Figure S1). However, the same program accurately predicted the secondary structures of the TPR helices (data not shown). As before, a formal attempt to align all 115 predicted TCH sequences by these programs failed to reveal a universal homology stretch in all TCHs; inclusion of the known TCH structures (Table 1) in the mix did not change the same outcome, ie, no consensus sequence was found (Figure S2). Thus, only the results of Clustal Omega, a commonly used and versatile MSA tool, are presented here (Figure S2).
Atomic-level interactions between TCH and TPR helices
Due to the lack of a consensus sequence in the TCHs, it was conjectured that if there is any interaction between TCHs and TPR, it might involve amino acid side chains that are not only non-identical but may also be positioned slightly differently in different proteins. Once they are properly identified, one could then ask whether there is a pattern among them. To this end, the PDB structure of these proteins was examined and the contacting residues were noted, as described in the “Methods” section. It was first tested whether the TPR-TCH area has a similar structure in different proteins of the same number of repeats, regardless of the nature or function of the protein and the amino acid sequence. Indeed, as shown (Figure 2), the 3TPR-TCH structures of PP5 and FKBP8, 2 otherwise different proteins, were highly superimposable with 82 atomic contacts that had a low Root-Mean-Square Deviation (RMSD) of 1.2 angstroms.
As PP5 dTCH (35 amino acids) is a relatively long helix, much longer than that of FKBP8 (13 amino acids), its C-terminal half extends outside the superimposition, thus suggesting that the extra length may play no role in the interaction with TPR. These results not only confirm that TPR domains of the same repeat number have analogous overall architecture2,3 but also show that the TCHs, regardless of their amino acid sequence or length, are positioned at a similar angle relative to their cognate TPRs, suggesting conservation of structural interaction over a similar length.
At this point, a detailed analysis of the interaction at the atomic level was in order. This was performed in 2 steps as described in the “Methods” section. First, I used the recently available and updated Protein Contacts Atlas 10 to identify all non-covalent contacts between the TCH and the other helices for the PDB numbers described above (Table 1). To prioritize, only the residues that showed more than 1 (ie, 2 and above) atomic contacts were considered for further analysis. The contacts were tabulated as an amino acid interaction matrix and observed in PyMol for both confirmation and presentation. Two examples are also structurally depicted, one for the single TCH in human PP5 (Figure 3) and the other for yeast ribosomal RNA biogenesis protein RRP5, which contained 1 uTCH and 2 dTCHs (Figure 4).

Interactions of different types of TCH and TPR helices in yeast RRP5. The analysis and presentation are similar to those of Figure 2 but were derived from the RRP5 structure (PDB 5C9S). (A) The six 34-amino acid TPR motifs are circled. Note the typical arrangement of a multi-TPR domain, particularly the 2 helices (A-helix and B-helix, unmarked) of each TPR at a typical angle and the gradual turn of the TPR units to form the superhelical bend. Also as shown, the adjoining non-TPR helices, such as dTCH1 and dTCH2, often structurally appear like continuations of the TPR. The TPR-closest TCHs (uTCH and dTCH1) are colored dark red, whereas the second dTCH is yellow. Panels B and C show side-chain interactions at the indicated ends of the 6TPR domain, respectively, between upstream TCH and TPR1 (B), and between the 2 downstream TCHs and TPR6 (C). The number of contacts between helices is also shown. TCH indicates TPR-companion helices; TPR, tetratricopeptide repeat.
The occurrence of the second downstream helix in this solved structure presented an opportunity to interrogate its interaction as well. In both proteins, several side chains of the aforementioned TCHs indeed made contacts with the nearest TPR helix. Specifically, expressed in position number and single letter amino acid codes, the residues A3, K6, and I13 of the dTCH of PP5 contacted the A-helix (Figure 3A), and the same residues, as well as residues K4, Y7, C10, N11, and V14, contacted the B-helix (Figure 3B).
Together, they contacted Y5 and M12 of the A-helix, and F17, L21, Y24, E25, and V28 of the B-helix. Interestingly, 3 residues of the TCH—namely, A3, K6, and I13—were shared by both A- and B-helices. Likewise, in PRP5, which is a 6TPR protein (Figure 4A), the first (proximal) dTCH contacted with B-helix and also with A-helix of the TPR6 (Figure 4C).
Interestingly, the second (distal) dTCH also made some contacts, albeit fewer, with its 2 closest neighbors, ie, the first dTCH and the TPR6 B-helix (Figure 4C). The uTCH also made contacts with its neighbors, ie, A-helix and B-helix of TPR1 (Figure 4B). A preponderance of hydrophobic residues in these interactions could be noted. In addition, as indicated with different views of the structure, the facing helical sides were involved in all interactions, for reasons of proximity and hydrophobic interactions, which are detailed below. Furthermore, significantly more contacts were made with helices that were closer, also for reasons of proximity.
Helical location of the TPR-TCH interacting residues
Once it was established that the TCHs indeed interact with adjoining TPR helices, I investigated the properties and locations of the interactive amino acids in all TPR proteins of known structure (Table 1), with the hope of finding a common interacting pattern, which would be functionally more meaningful than overall sequence homology. First, all amino acids involved in interhelical interactions were marked in their helical positions (Table 1, in red color), as described under the “Methods” section, that a significant number of residues of each helix (A and B) participated in interaction with the TCH. A greater number of contacts with the nearer helix, which was seen before in small scale, was also noticed in this expanded group. Thus, the A-helix, on the average, made only 20 contacts with the dTCH, whereas the B-helix made 58 contacts (Figure 5), nearly 3 times as much (also see Table 2). In a few extreme cases, exemplified by 4I17, the A-helix showed no interaction (0 contact) with the TCH, whereas the B-helix made 46 contacts (data not shown).

Number of contacts between TCH and the 2 helices (A-helix, AH; and B-helix, BH) of the nearest TPR. The contact numbers were obtained from Protein Contacts Atlas, as mentioned in the “Methods” section. Numbers from all proteins in Table 1 were added and the mean plotted (20 contacts between TCH and A-helix; 58 contacts between TCH and the B-helix). TCH indicates TPR-companion helices; TPR, tetratricopeptide repeat.
Energy of TPR-TCH interhelical interaction.
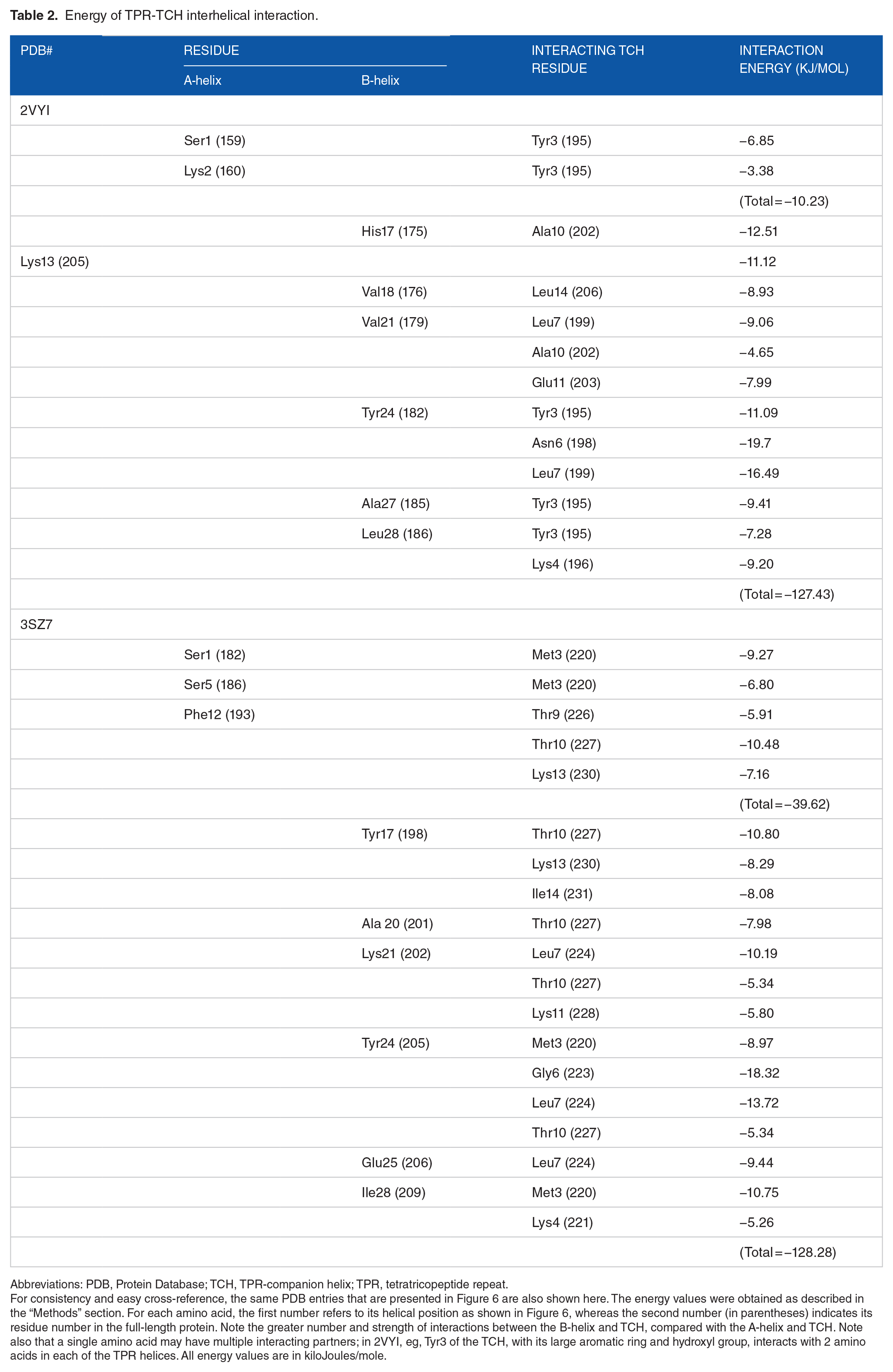
Abbreviations: PDB, Protein Database; TCH, TPR-companion helix; TPR, tetratricopeptide repeat.
For consistency and easy cross-reference, the same PDB entries that are presented in Figure 6 are also shown here. The energy values were obtained as described in the “Methods” section. For each amino acid, the first number refers to its helical position as shown in Figure 6, whereas the second number (in parentheses) indicates its residue number in the full-length protein. Note the greater number and strength of interactions between the B-helix and TCH, compared with the A-helix and TCH. Note also that a single amino acid may have multiple interacting partners; in 2VYI, eg, Tyr3 of the TCH, with its large aromatic ring and hydroxyl group, interacts with 2 amino acids in each of the TPR helices. All energy values are in kiloJoules/mole.
It was also noted that the contacting residues were located in spots that were separated in sequence by roughly 2 to 4 residues, which approximates the pitch of the alpha-helix (5.4 Å, equivalent to 3.6 residues). 11 The spread from 3.6 residues is likely because the interactions are often not one-to-one but rather a single amino acid of 1 helix makes contacts with multiple amino acids on the other helix. Moreover, side chains of different lengths and branches reach different distances to make contacts with slightly off-pitch residues. In other words, and as visualized before for PP5 and PRP5 (Figures 3 and 4), the distribution stems from the fact that residues occurring on a single face of a helix participate in interacting with another helix.
Examination of all interacting amino acid pairs revealed further details of the preferred sites of pairwise interaction. This is schematically illustrated with 2 arbitrarily chosen proteins (Figure 6), roughly representative of collective data (not shown) from all proteins in Table 1.

Example maps of the TPR-TCH interacting residues. Tetratricopeptide repeat domain protein structures were analyzed for helix-to-helix contacts as described in the “Methods” section, and 2 examples are shown. Each contacting cluster is indicated by a different colored line, which also shows that the same residue sometimes contacts more than 1 partner; eg, in both proteins, amino acid 21 of B-helix contacts both T9 and T10 of the downstream TCH. The same interactions are also detailed in terms of energy values in Table 2. As also mentioned in the “Results” section, the lines do not indicate the number of contacts between the 2 amino acids. The sequences of the helices are shown below the corresponding schematic diagram, and all interacting residues are indicated in red. The amino acid numbering system follows the schematic diagram in Figure 1. TCH indicates TPR-companion helix; TPR, tetratricopeptide repeat.
It should be mentioned that in this diagram, essentially all interactions involving 3 or more contacts between 2 amino acids have been indicated by a single connecting line. However, each line may represent widely different number of contacts between the 2 amino acids (ie, between different atoms of their side chains); these numbers were not displayed to avoid cluttering of the diagram. For example, in PDB 2VYI, the Y24 of the B-helix makes 7 contacts with L7 of the TCH, but S1 of the A-helix makes only 2 contacts with Y3 of the TCH, though they are both indicated by single lines. As shown in Table 2, the total energy of interaction between 2 helices is also related to their proximity, such that the TCH interacts much more strongly with B-helix than with the A-helix (−127.43 vs −10.23 kJ/mol in 2VYI, and −128.28 vs 39.62 kJ/mol in 3SZ7). The overall outcome is that the TCH interacts much more extensively and strongly with the B-helix than with the A-helix.
When a few amino acids dominate a particular position in a cohort of sequences, the multiple alignment may not reflect it, but a sequence logo plot generally does. Applied to the 34-amino acid long TPR sequences in the proteins of Table 1, sequence logo plot (Figure 7A) generated the signature TPR logo pattern, 4 essentially similar to the published plots for other TPR cohorts. 12

Sequence logo plot of amino acid abundance in TPR and TCH of 42 proteins of known structure (Table 1). The plot was performed as previously described 12 from 3TPR (A) and the adjoining Leu7-centric 11-mer peptide in the TCH (B), as described in the “Results” section. The major signature residues of TPR are indicated in black font, while the TCH-interactive residues are in red, showing that the 2 populations are different. TCH indicates TPR-companion helices; TPR, tetratricopeptide repeat.
Interestingly, most of the frequently occurring signature residues of TPR, notably 3A, Y4, G8, A20, and P32 (Figure 7A), tend to be absent in TCH interaction (Figures 6 and 7A, Table 2) which is due to their location on the other side of the helix or at different angles, as viewed in the available 3D structures (not shown). Thus, the TPR amino acids may consist of 2 functional classes, those involved in TPR function per se, and those engaged in making contacts with the TCH, possibly for the maintenance of structure and solubility.
Unfortunately, sequence logo plot could not be directly applied to full-length TCH sequences, as the operation requires sequences of identical lengths. In an effort to circumvent this hurdle, I concentrated on Y24 (Tyr24), located in the middle of the B-helix, as the single most frequently interactive amino acid in the TPR, which often interacted with Leu in the TCH, roughly at position 7 in independent numbering (Figure 6). Here and elsewhere, I have used “position 7” more as an eponym rather than an exact position count for all TCHs because, as mentioned before, the TCHs are of variable lengths, connected to the B-helix by spacers that are also of variable lengths. It is fair to assume that the proper alignment of the TCH with the B-helix is achieved by energetically optimal contacts (eg, Table 2) and not by absolute amino acid number count. All TCH sequences containing Y24-interactive Leu (from solved structures as well as corresponding locations in predicted structures) were sorted out and aligned by the Leu. The largest common denominator of the length of these peptides was 11 amino acids, with 5 amino acids on either side of the central Leu (Figure S3). A total of 38 such 11-mers were then subjected to sequence logo plot. As seen in the plot (Figure 7B), Leu stands out as the lone amino acid in the seventh position, evidently because the peptides were selected for its presence; reciprocally, no amino acid dominates in the other positions, further underscoring the heterogeneity in the sequence. Nevertheless, there were hints of some preference of aliphatic hydrophobic residues Leu, Ala, and Val at position 10, and basic residues Arg and Lys at position 12. Although not explored further, it is likely that this preference complements the interacting amino acids in the cognate positions of the B-helix.
Covariation of interacting amino acids
Preference of interaction between two amino acid residues is also reflected in their covariation, which was previously noted for interaction between two TPRs of the 3TPR modules. 4 In conducting a similar analysis between TCH and TPR, I considered not only Y24 of the TPR but also L24 and F24, the second and third most interactive amino acids in the same position. The frequency of their interaction with all 20 amino acids was then plotted in a 3D multi-bar plot (Figure 8A).

Covariation of position 24 of TPR and interacting position 7 of TCH. (A) The 3-dimensional multi-bar graph plots the frequency of all 20 possible amino acids (shown in single letter codes) in TCH that were found to make contacts with Y24, F24, or L24 in the TPR B-helix. The major interacting bars are indicated. (B) Visualization of interaction between Y24 and L7 (in PDB 2VYI) and between L24 and A7 (3 bonds, in PDB 2XEV), depicted in space-filling model. The actual position numbers of the amino acids in the full-length polypeptides are Y182-L199 (in 2VYI) and L98-A114 (in 2XEV). TCH indicates TPR-companion helix; TPR, tetratricopeptide repeat.
The plot revealed a strong preference of Y24 for L7 in the TCH, while L24 interacted with A7 and L7 nearly equally. Visualization of the interactive pairs Y24-L7 and L24-A7 in the PDB crystal structures authenticated the side-chain fit at 3D level (Figure 8B). The flat aromatic (phenolic) ring of the Tyr side chain and the relatively short side chain of Ala (-CH3) allowed space for the large (branched aliphatic) side chain of Leu, and the hydrophobicity of the side chains facilitated their contact.
Discussion and Conclusion
This project started with the goal of understanding whether or why the TCHs may act as a “solvation helix,”3,5 with the hope that bioinformatic analysis would find an interactive signature sequence, much like the TPR itself. However, extensive efforts with a variety of naturally occurring TPR proteins and TCH sequences established that a straightforward sequence consensus of TCH does not exist. Nonetheless, this study revealed important facets of the TCH, which can be summarized as follows: (1) the TCH constitutes a family of helices that are diverse in sequence, length, and position; (2) it makes significant contacts with at least 2 neighboring TPR helices, but more so with the closer helix, the so-called B-helix; (3) the contacts involve interaction between compatible side chains of proper length and hydrophobicity on the opposing helical faces; (4) many TPR proteins additionally contain a TPR-uTCH that also interacts with the proximal A-helix; (5) a minority of TPR proteins lack a recognizable TCH; and, finally, (6) most, if not all, TPR amino acids that are considered TPR signatures do not partake in interactions with the TCH, primarily due to their location on the TCH opposite side of the helix or at a farther distance.
It thus appears that the TPR-signature residues perform the roles that a TPR has been traditionally recognized for, such as protein-protein interactions, whereas specific non-signature residues of the TPR, lining 1 helical face, interact with the TCH. Reciprocally, the TCH acts as an amphipathic helix with side chains that are within atomic contact distance from the B-helix and the A-helix, but otherwise functions as a generic helix with no universally conserved signature residue or motif. The molecular mechanism of how the TCH facilitates solvation of the TPR domain remains unclear, but it is tempting to speculate that this is achieved by the masking of the hydrophobic residues of the last TPR helix, at the same time terminating the continuation of the TPR repeats once the requisite number of repeats has been achieved. The roles of the individual TPR-signature residues are also not precisely known, but many of them appear to serve in a structural role, including the formation of the extended helical core and turn stabilization. 3 It can be conjectured that the location of the TCH outside the TPR domain has led to a division of labor, which must have allowed the TPR domain the freedom to evolve its signature residues to achieve the optimal TPR-specific scaffold and function, while delegating the TCH to the solvation task. As stated at the beginning, the strongest evidence for the solvation function of TCH came from the studies of synthetic TPR peptides of consensus sequence; 4 nevertheless, this may also hold true for a naturally occurring full-length TPR protein, although this has not been specifically tested.
As briefly mentioned earlier, the TCH often structurally appears to be a continuation of the TPR series of helices, and like the alternate helices of the TPR, it is also antiparallel to the preceding TPR helix. Thus, it cannot be ruled out that in the proper context, the TCH may contribute to the concave substrate-binding groove of the TPR domain. A case in point is the IFIT5 protein, which contains a nearly circular superhelix vortex (named “eddy”) of a total of 24 helices, 14 of which are from 7 TPRs. 6 It is curious to note that a C-terminal deletion of IFIT5, which precisely lost the TCH sequence, could be expressed in soluble form, although it had lost the nucleotide-binding ability. 6 Thus, not all helices neighboring a TPR may act as solvation helix.
To explore if the TCH promotes a structural alteration in the interacting TPR helix, several PDB structures were closely examined in PyMol display, but no unusual interhelical angles or TPR helical architecture could be seen. Ideally, these structures should be compared with one in which the TCH is deleted, as in the C-terminal IFIT5 deletion mentioned above; 6 unfortunately, however, the structure of the ΔTCH-IFIT5 was not determined. As mentioned before, within a TPR, the A-helix and B-helix also make mutual contacts, but the structural effects of these contacts are also currently unknown.
Reciprocally, several TPRs appear to be exceptions to the solvation helix postulate, some of which are reported here (Figure S1). A notable TCH-free class is found in the 3TPR linkers that connect the FKBP (FK506-binding protein) and the CYN (cyclophilin) sequences in the dual-family prolyl isomerases of the domain structure FKBP-3TPR-CYN (abbreviated as FCBP). In this family of nearly 50 proteins, a long stretch of ∼60 amino acid residues is present between the last TPR motif (TPR3) and the downstream CYN domain. Although the structure of this region has not been experimentally determined, secondary and tertiary structure modeling did not reveal any helix.12,13 Nevertheless, these proteins, expressed recombinantly, are highly soluble.12,13 Thus, if a downstream companion helix is in fact needed for the solubility of 3TPR modules, the need may be context-dependent, such that in the absence of a companion helix, the presence of other domains in the protein or an abundance of hydrophilic amino acids in the TPR module itself may promote solubility. Indeed, the TPR3 modules of the FCBP 3TPRs are exceptionally rich in hydrophilic amino acids, specifically, the acidic amino acid, Asp, and basic amino acids, Arg and Lys,12,13 which may have obviated the need for a solvation helix. It is certainly beyond reason that every helix will need a downstream helix for solubility, because it conjures up the impossible scenario of an ad infinitum chain of helices in a linear protein.
Although I have drawn attention to the uTCHs, they could not be analyzed further due to the small number of solved structures available. Nevertheless, uTCH-TPR interactions were noted for 2 protein structures (Table 1, Figure 4) that also involved hydrophobic residues. Both happened to contain Pro at the end of the uTCH (Table 1); as proposed for specific Pro residues in the TPR helices (such as P32), 3 these prolines may serve as helix-breakers, ensuring termination of the uTCH for a clean start of the downstream A-helix. The second Pro in 3HYM, at the N-terminus of the A-helix, may act as helix initiator for the A-helix (Table 1).14,15 It remains to be seen if the uTCHs are capable of functioning as solvation helix in their natural context. Finally, for the sake of authenticity, attention in this study was focused on experimentally verified structures, mostly derived from X-ray crystallography; however, such structures are only a snapshot of a dynamic ensemble of structures that may be fully appreciated in such techniques as Molecular Dynamics simulation, which could offer a larger window of the TPR-TCH interaction. This could be an important area of future analysis.
Overall, the results presented here should generate interest in understanding the exact function of the under-appreciated TPR-companion helices that accompany the majority of TPR domains. If they are indeed specific promoters of solubility, properly matched helices may find applications in biotechnology to express recombinant TPR domains in soluble form3,5,13,16 without affecting the sequence or functionality of the TPR.
Supplemental Material
Supplementary_material_xyz20942018a1392 – Supplemental material for Protein Tetratricopeptide Repeat and the Companion Non-tetratricopeptide Repeat Helices: Bioinformatic Analysis of Interhelical Interactions
Supplemental material, Supplementary_material_xyz20942018a1392 for Protein Tetratricopeptide Repeat and the Companion Non-tetratricopeptide Repeat Helices: Bioinformatic Analysis of Interhelical Interactions by Sailen Barik in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
I acknowledge Dr Lynne Regan (Yale University, New Haven, CT, USA) for sharing the 3TPR sequence list that was used to retrieve the full-length parent protein sequences.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: No external funds were used for these studies. The article processing charge was paid for by the author’s personal fund.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SB, the sole author of this article, performed all aspects of the study, designing and conducting the analyses, and writing and communicating the article.
Data Availability Statement
All data generated in this study are included in this published article as stated.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.