
Abstract
The associations between proteins and diseases are crucial information for investigating pathological mechanisms. However, the number of known and reliable protein-disease associations is quite small. In this study, an analysis framework to infer associations between proteins and diseases was developed based on a large data set of a human protein-protein interaction network integrating an effective network search, namely, the reverse k-nearest neighbor (RkNN) search. The RkNN search was used to identify an impact of a protein on other proteins. Then, associations between proteins and diseases were inferred statistically. The method using the RkNN search yielded a much higher precision than a random selection, standard nearest neighbor search, or when applying the method to a random protein-protein interaction network. All protein-disease pair candidates were verified by a literature search. Supporting evidence for 596 pairs was identified. In addition, cluster analysis of these candidates revealed 10 promising groups of diseases to be further investigated experimentally. This method can be used to identify novel associations to better understand complex relationships between proteins and diseases.
Introduction
One of the most challenging aims in biological science is to understand the role of genetics in complex human diseases. To identify disease genes, a widely used method is genome-wide association studies (GWAS), which have been used to identify a number of polymorphisms that statistically correlate with complex diseases. In particular, GWAS attempt to detect associations between common single-nucleotide polymorphisms and common diseases. However, identifying such variants makes only a small contribution to explain disease occurrence.1,2 In addition, because of functional redundancy and because most proteins do not function in isolation, biological mechanisms are complicated and are usually studied from a network viewpoint.3,4 Studies of protein function could be enhanced by network-based analysis that has been shown to be useful to gain insight into biological mechanisms. Thus, network-based analysis could also help to find new protein functions important for a specific disease. To understand the relationship between genes and diseases, several studies discovered that partner proteins in a biological network tend to share common functions.5,6 In addition, the study of Lage et al 7 and other studies reveal that most of the causative genes of complex diseases are likely to reside in the same network modules, eg, pathways8,9 or subnetworks 10 of a given biological network. The study of Vanunu et al 11 performed network propagation methods, and other studies12–14 performed literature-based methods and network analysis to predict the association between genes and some specific diseases such as prostate and breast cancer, Alzheimer, and type 2 diabetes mellitus. A common method to infer a protein-disease relationship is to find shared known diseases between 2 neighboring proteins. However, several associations between proteins and diseases or between diseases and diseases are unrevealed and remain a challenging task.
To identify new disease proteins in a protein-protein interaction network, a common method such as a k-nearest neighbor (kNN) search was used in the study of Xu et al. 15 However, an alternative algorithm named the reverse kNN (RkNN) search that uses an inverse concept from kNN was invented. The RkNN was applied in several applications, eg, geographic information systems, databases, and business management.16–18 In biological networking, Ning et al 19 was the first study to use RkNN to find essential proteins in yeast. The concept of RkNN is reversed from kNN. Instead of finding neighboring proteins of a query protein, the RkNN considers which proteins have the query protein as their neighbors. With this concept, we could infer that those neighbor proteins are influenced by the query protein. If the set of neighbors are disease proteins, we could infer that the query protein tends to be a disease protein. The RkNN had been applied to disease studies and showed superior performance in a specific human disease, ie, inflammatory bowel disease. 20
This work is the first study that used RkNN method focusing on a large data set of human disease–related proteins to identify new protein-disease associations. We constructed a network-based analysis framework on a human protein-protein interaction network to identify novel protein-disease association pairs in several diseases. Using a network-based approach, one of the important factors is the use of reliable data sets to construct a network and to gain true information about disease proteins. Therefore, in this study, we used an integrative metadata of protein-protein interaction networks from the STRING database 21 with high confidence scores together with a well-defined disease protein data set from our gold standard that contains disease-related gene annotations from Online Mendelian Inheritance in Man (OMIM), UniProtKB, and the GWAS study from Phenotype-Genotype Integrator (PheGenI) databases. 22 In addition, to investigate the network, we used an RkNN search and tested statistically to infer protein-disease associations. Moreover, the results from our inference method were extended to find new relationships between diseases.
Materials and Methods
Network data and protein-disease annotations
Protein-protein interactions were collected from STRING database version 10. 21 This database contains both known and predicted protein-protein interactions and provides a confidence score for each pair of interactions based on the available evidence in several channels, eg, databases, co-occurrence, coexpression, gene fusion, and experiments. Thus, our human protein-protein interaction network was constructed using only reliable interactions having high confidence scores of more than 900 as a weighted network. Finally, the network contains 17 880 proteins and 203 319 interactions. About 87% of these selected interactions have evidence in the database channel from STRING. The evidence in the database channel was aggregated from KEGG pathway database and then was asserted by human expert curators. Therefore, these interactions in our analysis include both the physical and functional interactions. For our gold standard of disease proteins, the well-defined disease-gene pair data set from the study of Menche et al 23 was used. Those authors collected disease-gene pair annotations from OMIM (www.omim.org), UniProtKB/Swiss-Prot mapped by Mottaz et al, 24 and the GWAS data from the PheGenI databases (https://www.ncbi.nlm.nih.gov/gap/phegeni). 22 Different disease nomenclatures from different sources were merged into a single-standard vocabulary using the Medical Subject Headings ontology (MeSH; www.nlm.nih.gov/mesh/). Genes and corresponding proteins were mapped. There were 299 diseases with 3173 proteins after filtering out diseases with less than 20 associated genes in our gold standard.
Reverse kNN search
A reverse nearest neighbor search is a method to find node(s) for which a query node is its/their neighbor. Normally, it has a parameter k to indicate the number of considered nearest neighbors of the query node. Therefore, we called it an RkNN search in general. The concept of the search is to find the neighboring nodes that are influenced by a query node. For a protein-protein interaction network, the RkNN search was employed for finding proteins that are influenced by a query protein. The weights of our protein-protein interaction network are the confidence scores from the STRING database. Therefore, the distance of each edge connected between 2 proteins is an inversion of the confidence score between 2 proteins. The formulation of the RkNN search is as follows.
Let distance be the distance between 2 proteins and let P be a set of proteins in a network. The k-nearest neighbors of a protein q is the k-closest proteins to q. It is defined by kNN(q) such that

The set of RkNN of query protein q is defined as follows:

In other words, p in the set of RkNN(q) is a protein that is influenced by protein q. Therefore, with the same parameter k, RkNN and kNN of a query protein provide different sets of proteins. Instead of simply finding k-nearest proteins to a query protein such as kNN search, RkNN attempts to identify a set of proteins that the query protein is their kNN. Therefore, the RkNN always provides a smaller set of influenced proteins, whereas the kNN provides the set of k-nearest (or closest) proteins to a query protein. With a larger list of nearest neighbors by kNN, some irrelevant neighbors that might not affect the query protein may be included and add some noise to the precision of the prediction.
Statistical test for inferring protein-disease associations
After the RkNN gives a set of proteins influenced by a query protein, the enrichment test is performed on this set of influenced proteins to their known diseases according to our gold standard. Protein q whose RkNN proteins are statistically overrepresented with a disease is inferred as related to that disease. This statistical test was performed using the 1-sided Fisher exact test, and the P value criterion was defined as .01. In this study, a set of RkNN proteins of a query protein was examined for all 299 diseases, and all proteins in the network were used as query proteins. Finally, we obtained a list of protein-disease pairs. To measure the performance of protein-disease association identification, precision was calculated as the ratio of the number of true protein-disease pairs detected to the number of protein-disease pairs identified.
Clustering method
To identify highly connected and dense regions in the protein-disease association network, we employed the clustering algorithm MCODE, 25 which is a plug-in of Cytoscape software. 26 MCODE calculates the local neighborhood density of a protein in the network and assigns a value to the protein. Clusters are constructed around the top-weighted protein nodes by iteratively adding high-scoring protein nodes to the cluster. Only dense clusters are selected for the final set of partitions. 27 In this study, we used a default node cutoff value of 0.2, a K-core value of 2, and the Haircut algorithm to exclude nodes with a low degree of connectivity from the cluster. The score of a cluster was computed as a product of the subgraph density and the number of nodes in the cluster.
Results
To find a relationship between an unknown disease-related protein and diseases, we used the basic concept of disease inference based on neighboring proteins. Briefly, the concept hypothesized that proteins that are directly connected in a protein-protein network could share a common disease. With this hypothesis, a computational framework was constructed to analyze disease proteins using the interaction network. An overview of this framework is shown in Figure 1. To predict protein-disease associations, a network of protein-protein interaction was constructed. With this network, a set of influenced proteins of a query protein was discovered by the RkNN method. Integrating with information from known disease proteins, we could infer groups of diseases that might be related. The validation of these relationships could be performed by text-mining PubMed.
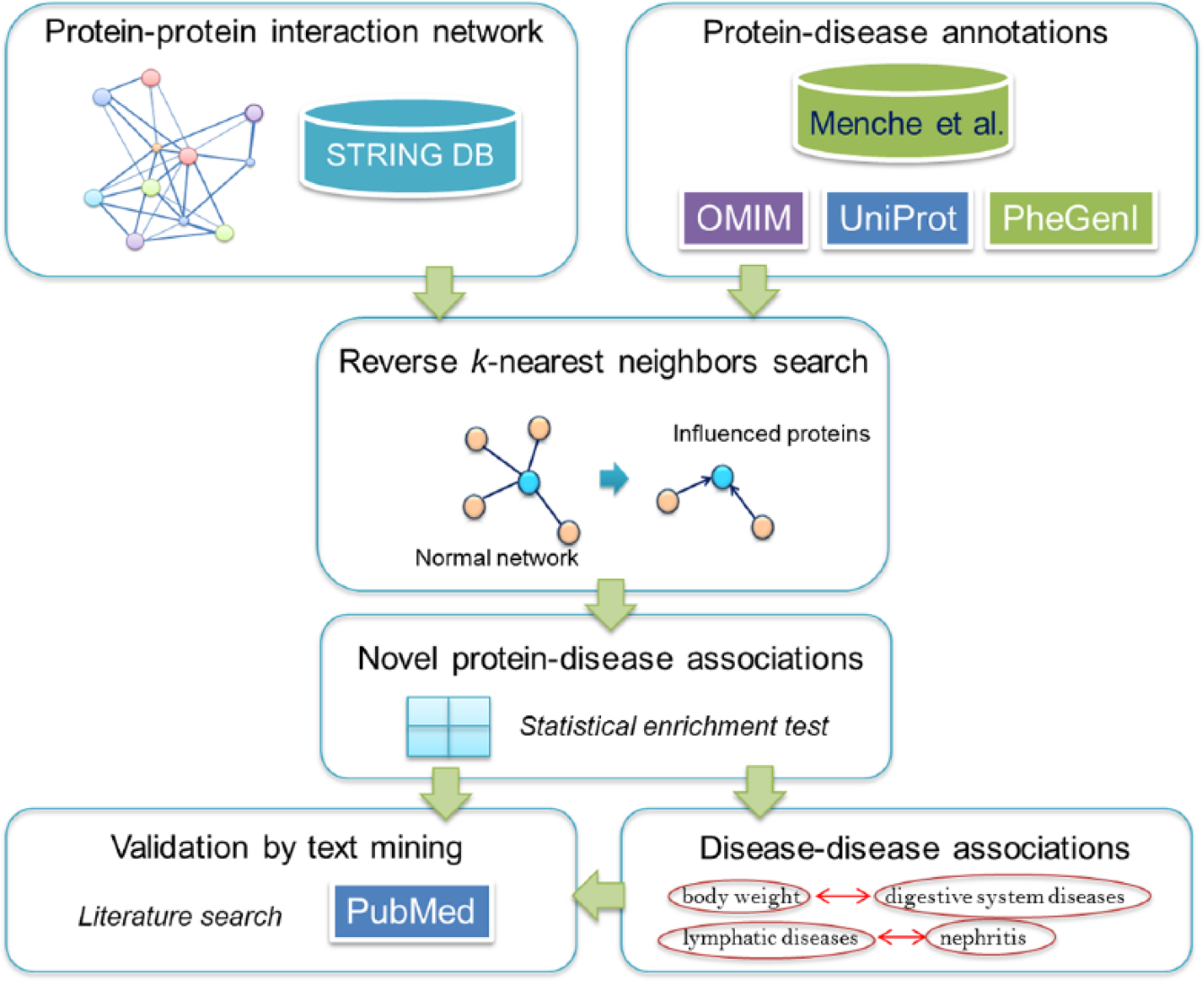
Overview of the method. The framework starts by constructing a protein-protein interaction network using the information from the STRING database. 21 Integrating the protein-disease annotations from Menche et al, 23 we applied the reverse k-nearest neighbor algorithm to the network for identifying influenced proteins for each protein in the network. Then, an enrichment analysis of the diseases that were significantly related to the influenced proteins was undertaken, and the association between each protein and each disease was inferred. Later, the protein-disease pair candidates were used for finding disease-disease associations. Finally, all candidate pairs, either protein-disease pairs or disease-disease pairs, were validated by text mining the PubMed database.
Protein-protein interaction network to protein-disease associations
A protein-protein interaction network for humans was constructed using the information from the STRING database. 21 Only interactions with high confidence scores of more than 900 were collected and combined to yield a network of 10 573 proteins and 203 319 interactions. This network follows the scale-free network with the exponent value of the fitted power-law distribution of 1.5521 (see Figure 2) and has the basic network properties shown in Table 1. Notice that this network is quite dense with hubs (high-average degree nodes of 22.7426) and low clusters (low-average clustering coefficient of 0.2673). This indicates the robustness of the network with perturbation and that the communication in local networks depends on neighboring nodes rather than on the connections among neighbors. Thus, inferring information from the neighboring nodes is of great value. Moreover, finding such a dominated neighboring node would be important, as we did in this study using an RkNN search.

Degree distribution of our protein-protein interaction network is scale free.
Network properties of the constructed protein-protein interaction network.

The RkNN finds a set of proteins influenced by a protein of interest (see “Materials and methods”). Associated diseases of these influenced proteins were sought using an enrichment test with the Fisher exact test. If the influenced proteins were enriched with P < .01 on a set of disease-related proteins, that disease should also be related to the query protein. A set of influenced proteins of a query protein was tested for all sets of proteins of 299 diseases. Finally, only the diseases for which the related proteins were enriched by the influenced proteins were selected to be related to the query protein. The RkNN search was performed with different values of parameter k ranging from k = 1 to k = 30. For each parameter k, each protein in the network was used as a query protein and the list of predicted associations was produced. To find an optimal parameter k, the precision of our predictions was calculated (see “Materials and methods”) using known disease proteins from the gold standard. We found that when k = 1, the RkNN method yielded the best precision of 0.36. The precision gradually declined when the value of k increased. With this optimal parameter k, we obtained 1502 candidate pairs of which 546 pairs were found in the gold standard.
Interestingly, the optimal k parameter equals 1 (k = 1) with the RkNN search. This result occurs due to the special characteristics of the searching method. With k = 1, it is possible to find more than one influenced protein. In contrast, the standard kNN always gives a single protein when selecting a parameter k = 1. Therefore, choosing parameter k = 1 in an RkNN search could obtain proteins that are exactly related to the query protein. The other issue concerns the precision of our method. We found that the number of known protein-disease pairs is very small. With our gold standard, we have 29 775 protein-disease pairs. Considering overall 299 diseases and a total number of 10 573 proteins in our network, we have all possible 3 161 327 protein-disease pairs. That means, if we randomly detect 100 protein-disease pairs, there will be approximately one (100 × 29 775/3 131 552) protein-disease pair that might be the true protein-disease relationship. This indicated that random selection yields a precision of only 0.0095 or 1%.
Protein-disease association candidates
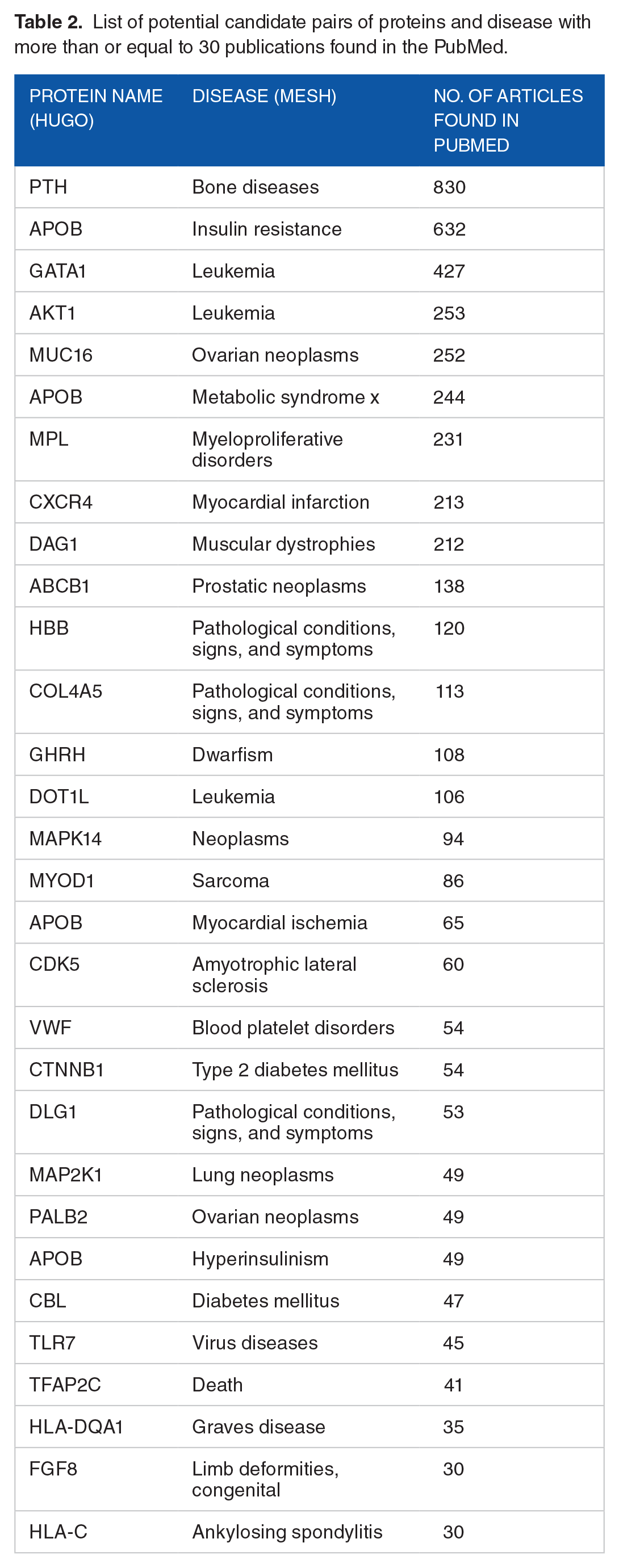
To find further supporting evidence for our predictions, we validated our results using a text-mining search. Each predicted pair of a disease protein and a disease name was queried on PubMed at the National Center for Biotechnology Information (NCBI) database (www.ncbi.nlm.nih.gov). The query keywords consisted of a protein symbol denoted according to the HUGO Gene Nomenclature Committee (www.genenames.org) and a disease name nomenclature from the MeSH database. Precision was calculated by counting the number of predictions that found at least one report in the NCBI database divided by the number of predictions. Interestingly, the RkNN search found literature support for 596 pairs from all 1502 candidate pairs (giving a precision of 596/1502 = 0.3968). Combining the results from our gold standard and the literature search, we found 316 predicted pairs in both the gold standard and the literature search (see Figure 3). However, 230 of 1502 predicted pairs were found in the gold standard but not found in the literature search. For the pairs found in the literature search but not in the gold standard, we obtained 280 predicted pairs, but 676 predicted pairs were not found in both the gold standard and the literature search. Table 2 shows the list of potential protein-disease association candidate pairs that were not found in the gold standard but had more than 30 reports in PubMed. The complete list of all candidate pairs can be found in Supplementary Table S1.
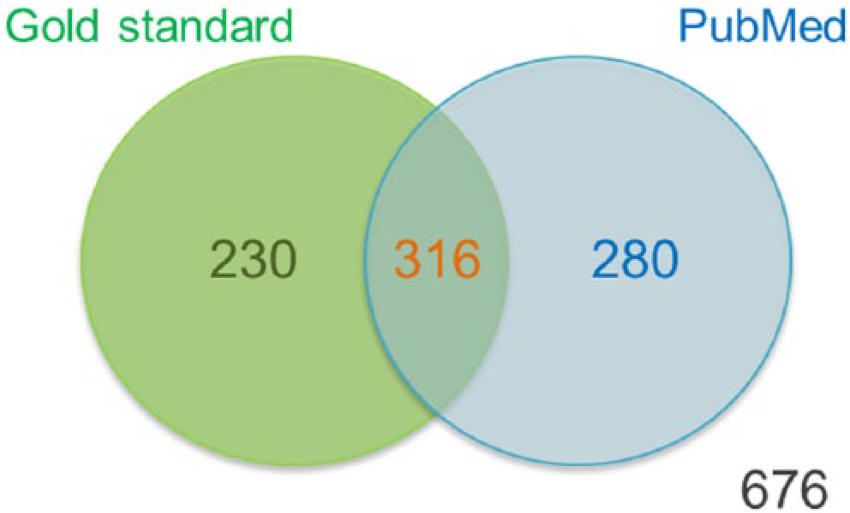
Venn diagram of the number of protein-disease association pairs.
List of potential candidate pairs of proteins and disease with more than or equal to 30 publications found in the PubMed.
Effectiveness of RkNN over the standard searches
To demonstrate the effectiveness of the RkNN search in finding only influenced proteins among neighbors of a query protein rather than the standard kNN search, the same process was undertaken using the kNN search instead of the RkNN. The set of neighbor proteins of a query protein were used for performing the enrichment test. With the same P value cutoff and the same gold standard set, the precision of each parameter k was calculated. The comparison of the RkNN and the kNN results is shown in Figure 4. Notice that for all ranges of parameter k, the RkNN outperformed the kNN. The highest precision when performing the kNN when k = 4 was 0.21, which is lower than the precision when performing RkNN. For the literature validation, we obtained a higher precision of 0.3968 for the RkNN search compared with the kNN search that yielded a precision of 0.2752 (found literature of 2435 predictions from all 8849 predictions). Furthermore, to demonstrate how important it is to use a reliable interaction data set, we conducted the same procedure on a random interaction network. This network was generated to contain the same number of proteins and interactions and to be a scale-free network as well. Both an RkNN search and kNN searches were applied to the random network. This scenario was repeated 5 times and yielded very-low-average precisions (less than 0.007) for both search methods for all values of parameter k. This precision value of 0.007 from this random experiment corresponds to the random selection, as mentioned above, that yielded a precision of 0.0095. Therefore, our method, with a precision of 0.36 for a large set of unbalanced data, as in this case, is very high compared with random detection.

Performance of identifying protein-disease associations using RkNN and kNN methods. The barplots illustrate the precision of protein-disease association predictions by the RkNN and kNN methods. The precisions of both methods are compared by varying parameter k from 1 to 30. The fractions of the number of true protein-disease association detected to the number of identified protein-disease association are presented on the top of each bar. kNN indicates k-nearest neighbor; RkNN, reverse k-nearest neighbor.
Robustness of the method to the interfered network
To validate the robustness of our method, we investigated the effective of the predictions when the network was perturbed. The original network was interfered by removing important nodes in the network and then used the interfered network to perform our analysis framework resulting in the precisions of their predictions. These important proteins were defined as proteins that have high degree value. The first interfered network was constructed by removing 366 proteins whose node degrees were more than 300. The second and the last experiments were performed on the interfered networks by removing 443 proteins having node degrees more than 200 and 1111 proteins having node degrees more than 100, respectively. The same tendencies of the results as the original network were found for these 3 experiments. The capability of our method with RkNN outperformed the method with kNN for all values of parameter k for the first and the second experiments. For the last experiment, when removing proteins having node degree more than 100, our method with RkNN outperformed the method with kNN at only for small values of parameter k and the rest of the performances were similar when the values of k became larger. These results illustrated the robustness of our method with RkNN on the interfered network. Figure 5 shows the results of these 3 experiments.

Performance of identifying protein-disease associations using RkNN and kNN methods on the interfered network. (A)-(C) show performances of the methods on the interfered network by removing proteins that have node degree more than 300, 200, and 100, respectively. kNN indicates k-nearest neighbor; RkNN, reverse k-nearest neighbor.
Clusters of the protein-disease association network
The protein-disease association network was constructed using the resulting protein-disease association candidates. This network consists of 633 nodes of proteins and 246 nodes of diseases and 1502 interactions between proteins and diseases. The MCODE clustering algorithm 25 (see “Materials and methods”) was applied to the network to find highly interconnected subgroups. We identified 10 interesting clusters that contain 23 proteins and 24 diseases, whereas the other proteins and diseases were isolated separately. The complete list of the clusters is shown in Table 3. The complete picture of these 10 clusters is shown in Figure 6.
List of 10 clusters consisting of clustering score, the number of nodes and edges, and the lists of proteins and diseases in each cluster.

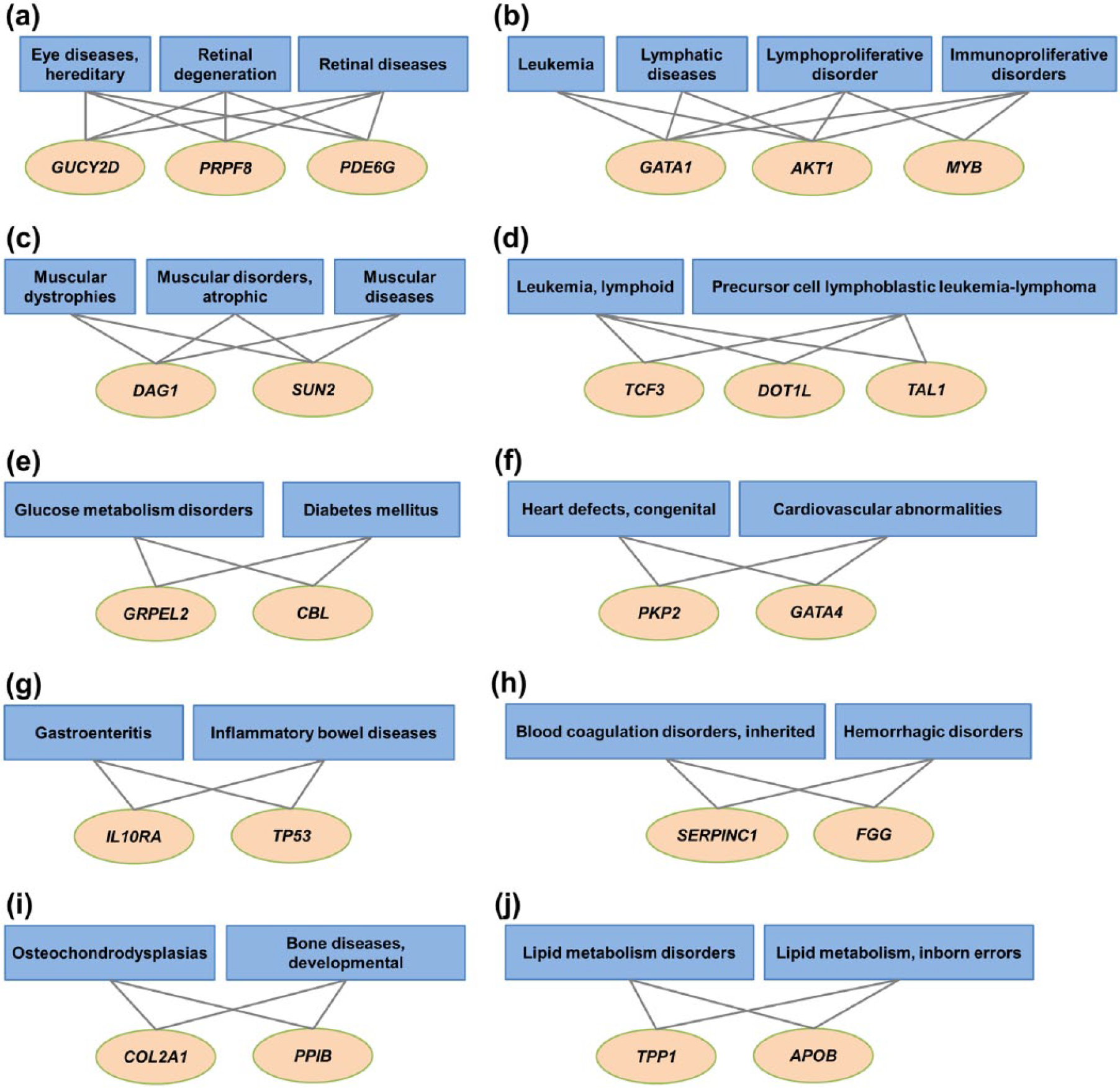
Clustering results. (A)-(J) present ten promising clusters found in the protein-disease association network.
The cluster with the highest score of 3.6 consisted of 6 nodes and 9 interactions. These 6 nodes comprised 3 proteins—pre-mRNA processing factor 8 (PRPF8), retinal guanylate cyclase 2D (GUCY2D), and phosphodiesterase 6G (PDE6G)—and 3 diseases—(1) retinal diseases, (2) retinal degeneration, and (3) eye diseases, hereditary. Each protein in this cluster links to all 3 diseases related to retinal and eye disease. GUCY2D is well known to be related to retinal disease, retinal degeneration, and eye diseases. Mutations in GUCY2D are the cause of inherited retinal degeneration.28,29 There is evidence that mutations in PRPF8 and PDE6G are related to retinal diseases and retinal degeneration.30,31 However, we could not found a relationship with hereditary eye diseases.
The second cluster yielded a cluster score of 0.333. This cluster comprised 7 nodes, containing 3 proteins and 4 diseases, and 10 interactions. The 3 proteins were as follows: (1) GATA-binding protein 1 (GATA1), (2) AKT serine/threonine kinase 1 (AKT1), and (3) MYB proto-oncogene, transcription factor (MYB). The 4 diseases were as follows: (1) lymphoproliferative disorders, (2) lymphatic diseases, (3) leukemia, and (4) immunoproliferative disorders. In this cluster, AKT1 and GATA1 interacted with all 4 diseases. AKT1 regulates many processes, including metabolism, proliferation, cell survival, growth, and angiogenesis.32–34 It is activated in acute lymphoblastic leukemia 35 and is a promising target for combination therapy in acute myeloid leukemia. 36 Although we could not find literature to support a direct relationship between AKT1 and immunoproliferative disorders, we found that AKT1 is implicated in X-linked lymphoproliferative disease type 1 (XLP1), a rare inherited immunodeficiency disorder. 37 GATA1 is a crucial regulator of megakaryocyte differentiation. Deficiency of GATA1 leads to megakaryoblastic leukemia. 38 In addition, GATA1 transcription factor is required for murine dendritic cell (DC) development. Dendritic cell–specific GATA1 knockout mice had lower DC migration toward peripheral lymph nodes. 39 Similar to AKT1, we could not find a direct relationship between GATA1 and immunoproliferative disorders. MYB was predicted to be connected to lymphoproliferative and immunoproliferative disorders. MYB was found to play a key role in the regulation of hematopoiesis 40 that is possible to relate to these 2 diseases. Lymphoproliferative disorders are related to conditions in which lymphocytes are excessively produced and they present as a subclass of immunoproliferative disorders in the MeSH database.
The third cluster, with a score of 3.0, comprised 5 nodes and 6 interactions. Sad1 and UNC84 domain containing 2 (SUN2) and dystroglycan 1 (DAG1) were the 2 protein nodes. The 3 diseases in this cluster were as follows: (1) muscular disorders, atrophic, (2) muscular diseases, and (3) muscular dystrophies. SUN2 is one of the SUN proteins that are members of the linker of nucleoskeleton and cytoskeleton (LINC) complex. The LINC complex and the nucleoskeleton are essential for nuclear movement and positioning in the muscle cell. 41 Variants in SUN1 and SUN2 were identified in patients with Emery-Dreifuss muscular dystrophy. 42 However, evidence of an association of the SUN protein to muscle atrophy was not found. Mutations of DAG1 and at least 17 other genes interrupt the extracellular matrix receptor function of dystroglycan that is a glycosylated basement membrane receptor involved in maintaining processes of skeletal muscle. 43 Its mutation was also found in patients with mild muscular dystrophy and asymptomatic hyperCKemia. 44 It was also found that abnormal glycosylation of α-dystroglycan is a common pathomechanism of Fukuyama-type congenital muscular dystrophy, muscle-eye-brain disease, and Walker-Warburg syndrome. 45 To summarize these above details, we concluded the status based on literature of the predicted associations in Supplementary Table S2.
Disease-disease association candidates
In addition to identifying protein-disease associations, we also inferred disease-disease associations from our protein-disease candidate pairs. From our predictions of a relationship between a query protein and diseases, if that query protein is already known its related disease, we could infer a relationship between the disease of the query protein and its predicted disease. With the disease-disease relationship predictions, we identified 6142 disease-disease pairs. Interestingly, we found that 67% (4120 predictions) of our predictions have literature evidence in PubMed. The complete list of predicted disease-disease relationships, including information from literature searches, is shown in Supplementary Table S3.
Conclusions and Discussion
This study developed an analysis framework to infer associations between proteins and diseases based on a protein-protein interaction network with an integration of disease-related genes. The RkNN search was employed to find a set of influenced proteins of a query protein. Protein-disease associations were then identified statistically with known disease proteins. Our framework with an RkNN search outperformed a standard nearest neighbor search with a much higher precision. All protein-disease pair candidates were verified by literature searches and we found literature support for 596 pairs. It is to note that the number of literature supporting predicted pairs could be changed depending on time. However, this is irrelevant because the number of found literature for a pair that really has an association should be significantly higher than the number of found literature for a pair that is not involved. The results from the cluster analysis of these candidates revealed 10 promising groups of diseases, eg, a group of eye and retinal diseases, a group of lymphatic, lymphatic, and immunoproliferative diseases, and a group of muscular dystrophies. These clusters can be used to be further investigated experimentally. Furthermore, not only did we infer protein-disease associations, but we also extended our association results to find disease-disease associations. This investigation resulted in more than half of all predicted disease-disease association pairs that were listed and verified with publications. An issue concerning results of disease-disease association prediction is about common etiology. We should consider the single etiology of disease pairs to avoid artifacts. Unfortunately, a database of etiology for a large set of human diseases does not exist at the moment.
We examined our method using another database as our gold standard. With this experiment, we selected DisGeNET database, 46 one of the largest databases of genes and variants involved in human diseases. This database integrated data from text-mining method, information on Mendelian and complex diseases, and data from animal disease models. The results showed the same tendency that our method with RkNN outperformed the method with kNN. Performing our framework with DisGeNET database could show the good capability of our method. However, the precisions of the methods with RkNN and kNN were slightly reduced. We gained the highest precision of 0.22 and 0.14 for our method with RkNN and kNN, respectively. These declined precisions might be a result of using noise data as our gold standard. This database contained both experimental and computational information that might have some irrelevant data. The results are shown in Supplementary Figure S1.
One important process in our method is the statistical enrichment test. With this step, we need to be cautious of the number of disease-related proteins. The number of proteins to perform the enrichment test should not be too small to avoid statistical bias. In addition, our method took more computational time than standard method. To find influenced proteins, we first need to know the set of kNN of a query protein. Therefore, this RkNN method is a further step after we obtained results from the kNN search. That means, it certainly took more computational time than kNN method. However, it is worth doing more tasks to increase the precision of the method.
In conclusion, based on an integration of protein-disease data, a protein-protein interaction network, and an effective RkNN search method, novel protein-disease associations can be identified effectively. Our method is efficient to identify protein-disease associations on an interaction network that gives us opportunities to discover common pathological causes and mechanisms in different diseases. It might be useful for disease diagnosis and treatment suggestions for one disease based on other related diseases. In addition, it can be generalized to other association studies to enhance knowledge in biomedical science.
Footnotes
Acknowledgements
The authors thank the peer reviewers for their helpful comments that have improved the manuscript.
Peer review:
Two peer reviewers contributed to the peer review report. Reviewers’ reports totaled 794 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by King Mongkut’s University of Technology North Bangkok. Contract number KMUTNB-GEN-59-22.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AS conceived and designed the experiments, analyzed the data, and wrote the first draft of the manuscript. AS and KP contributed to the writing of the manuscript, agree with manuscript results and conclusions, jointly developed the structure and arguments for the paper, made critical revisions, and approved final version. Both authors reviewed and approved the final manuscript.
Disclosures and Ethics
As a requirement of publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.