
Abstract
Citronella (Cymbopogon winterianus) is one of the richest sources of high-value isoprenoid aromatic compounds used as flavour, fragrance, and therapeutic elements. These isoprenoid compounds are synthesized by 2 independent pathways: mevalonate pathway and 2-C-methyl-
Introduction
Cymbopogon winterianus (citronella) is an aromatic grass having a large repository of terpenoid compounds (essential oils). This crop is grown for commercial and industrial purposes due to its essential oil (citronella oil) used in perfumes, as flavour additives, and even as pharmaceutical products. 1 This crop is mostly grown in tropics and subtropics of Asia, America, and Africa. 2 Traditionally, citronella oil is also well known for its insect repellent quality and antifungal nature, 3 in addition to its medicinal use against various diseases.4–6 In a recent study, citronellal and linalool, 2 constituents of citronella oil, were reported to have strong antifungal activity against several species of Aspergillus, Penicillium, and Eurotium. 7 Moreover, citronella oil is used to control muscle spasms, expel worms from intestines, increase urine production (as a diuretic), and increase appetite. 8
The Java citronella oil has constituents such as limonene, geraniol, elemol, geranyl acetate, α-cadinol, citronellol, and citronellal, which account for 1% of fresh weight of leaf.
9
The processors for all these isoprenoid compounds are 2 common 5-carbon molecules, ie, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). In higher plants, both IPP and DMAPP are synthesized by 2 non-related biosynthetic pathways, ie, mevalonate (MVA) pathway and 2-C-methyl-
In view of its significance in isoprenoid metabolism, HMGR gene has been extensively studied in many plant systems, namely, Euphorbia pekinensis, 27 Catharanthus roseus, 28 Taxus media, 29 Ginkgo biloba, 30 Corylus avellana, 31 Cucumis melo, 32 Michelia chapensis, 33 Salvia miltiorrhiza, 34 Eucommia ulmoides, 35 Withania somnifera, 36 and Centella asiatica. 37 Cymbopogon winterianus is an important medicinal aromatic plant, but very few enzymes involved in the terpenoid biosynthesis have been sequenced and characterized till date. The enzyme HMGR, which is a vital enzyme and catalyses the initial steps of IPP biosynthesis, has not been characterized from C winterianus till now. This article reports the isolation, characterization, and tissue-specific expression analysis of CwHMGR (HMGR gene of C winterianus) as a first step in understanding monoterpene biosynthesis pathway in C winterianus. The result from our study is expected to shed penetrating insights into the regulation of secondary metabolite biosynthetic pathways by ubiquitous enzyme CwHMGR in an important aromatic medicinal plant, citronella.
Methods
Sequencing of full-length CwHMGR by rapid amplification of complementary DNA ends (RACE)
The partial sequence of CwHMGR was obtained from our ongoing C winterianus whole transcriptome project (Bioproject ID: PRJNA263976) but was truncated from 5′ end. Thus, gene-specific primers were designed manually for rapid amplification of complementary DNA (cDNA) ends by polymerase chain reaction (RACE-PCR) (5′-ACGGGATCTTCCCTGCCACGA-3′) to obtain full-length coding sequence. The 5′ RACE-PCR was performed using the GeneRacer Kit (Invitrogen, Carlsbad, CA, USA) as per manufacturer’s instruction. The 5′-RACE-PCR product was sequenced in ABI3500 sequencer (Applied Biosystems, Foster City, CA, USA) and aligned with the partial sequence to obtain the full-length HMGR gene sequence (GenBank accession no. KM504513). The DNA sequence thus obtained was conceptually translated to primary protein sequence using ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). 38
Primary sequence analysis
The primary protein sequence (GenBank accession no. AIZ09174) was analysed with various bioinformatics tools, namely, InterProScan, 39 SMART (Simple Modular Architecture Research Tool; http://smart.embl-heidelberg.de), 40 Conserved Domain Database (CDD; https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml), 38 and Pfam database (http://pfam.sanger.ac.uk/), 41 to deduce the protein family HMGR gene and to explore the domain arrangement within the protein. The membrane protein topology was predicted in TMHMM server using a method based on hidden Markov model 42 (http://www.cbs.dtu.dk/services/TMHMM/). The ProtParam tool of ExPASy proteomic server 43 was used to analyse physicochemical property of the protein based on primary sequence of CwHMGR. The CONCORD Web server (http://helios.princeton.edu/CONCORD/) 44 was used to predict the secondary structure of the protein.
Phylogenetic analysis
Primary sequence of CwHMGR was subjected to Basic Local Alignment Search Tool (BLAST) search against non-redundant (NR) database of National Center for Biotechnology Information (NCBI). The sequences showing significant homology (identity cutoff, >75%; query coverage, >80%; and E value ⩽ 0) were aligned using ClustalW tool 45 and represented using ESPript wave tool. 46 The 2-dimensional unrooted phylogenetic tree was constructed using the set of aligned sequences by implementing the neighbour-joining method in MEGA v6.1.0. 47 A bootstrap replication of 1000 was used as it is preferable for estimating the reliability of the phylogenetic tree.
Homology modelling of CwHMGR
To generate a homology model of CwHMGR protein, Domain Enhanced Lookup Time Accelerated BLAST (DELTA-BLAST) was performed against Protein Data Bank (PDB) to select the suitable templates. To cross-check the reliability of templates identified through DELTA-BLAST program, we used fold-recognition servers for template identification which includes 3D-Jury, 48 Pcons.net, 49 and Geno3D. 50 The template showing maximum identity and highest percentage of query coverage according to both fold-recognition servers and DELTA-BLAST was finally chosen for the construction of 3-dimensional (3D) model of CwHMGR using Discovery Studio 3.5 (Accelrys, Inc. San Diego, CA, USA). Among large number of rough models, the model with the lowest Discrete Optimized Protein Energy (DOPE) score was selected as best and further subjected for loop and side-chain refinement using Discovery Studio 3.5.
Model validation
The predicted loop refined model of CwHMGR was subjected to model validation process using PROCHECK tool 51 embedded in Structural Analysis and Verification Server (SAVES) server. The PROCHECK tool quantifies the amino acid residues in the available zones of Ramachandran plot and assesses the stereochemical quality of the model. The ProSA 52 tool was used to assess the energy profile of the CwHMGR model. The root mean square deviation (RMSD) of the corresponding Cα-atom pairs of both the model and template was calculated using iPBA 53 Web server. Finally, the refined model was subjected for further refinement by molecular dynamics (MD) simulations.
MD simulation of CwHMGR
The MD simulation of CwHMGR was performed with GROMOS96 43A1 force field in GROMACS 4.6 package. The protonation states of all ionizable residues in the modelled protein were set to their normal states at pH 7.0. During the MD simulations, all atoms of the protein were surrounded by an octahedron water box of SPC3 water molecules that extended 1.2 nm from the protein, and periodic boundary conditions were applied in all directions. The system was neutralized using equal number of counter ions (Na+/Cl−) with a concentration of 0.15 M. Energy minimization was performed using steepest descent algorithm for 2000 steps. A 500-ps position-restrained MD simulation was performed for the system followed by 30-ns MD simulations at constant pressure (1 atm) and temperature (300 K). The electrostatic interactions were calculated by the particle mesh Ewald (PME) algorithm and all bonds were constrained using LINear Constraint Solver (LINCS) algorithm. A cutoff value was set for long-range interactions, including 0.9 nm for van der Waals and 1.4 nm for electrostatic interactions, using the PME method. The snapshots were collected at every 1 ps and stored for further analysis. The system stability and differences in the trajectories, RMSD, root mean square fluctuation (RMSF), and the energies of the system were analysed using Xmgrace.
Tissue-specific expression profiling by real-time and quantitative reverse transcription-polymerase chain reaction
The differential expression of the CwHMGR gene in 3 different tissues of citronella (leaf sheath, leaf, and root) was analysed by semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR) and quantitative real-time PCR. The primers were designed by primer3 (http://primer3.ut.ee/), 54 with 60% to 65% of GC content and melting temperature ranging from 60°C to 65°C. The first strand cDNA was synthesized using PrimeScript 1st strand cDNA Synthesis Kit (Takara Clontech, Japan) as per manufacturer’s instruction. The 10-µL reaction mixtures with 125 ng first strand cDNA, 1× Taq polymerase buffer (Genei, Bangalore, India), 1U Taq polymerase (Genei), 10 pmol both forward and reverse primers (Sigma, St. Louis, MO, USA), and 5 mmol deoxynucleotide triphosphate mixture (Invitrogen) were subjected to semi-quantitative RT-PCR in GeneAmp thermal cycler (Applied Biosystems, Foster City, CA, USA). The amplification conditions were 5 minutes at 94°C, followed by 35 cycles of 1 minute at 94°C, 1 minute at 67.5°C, 1 minute at 72°C, and a final extension for 10 minutes at 72°C. The amplified products were visualized in 2% agarose gel stained with ethidium bromide. In this study, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as reference. The relative expression of HMGR gene in the same 3 tissues was checked with quantitative real-time PCR by ΔΔCT method 38 on StepOnePlus Real-Time PCR System (Applied Biosystems) using the SuperScript III Platinum SYBR Green One-Step qRT-PCR with ROX Kit (Invitrogen) according to manufacturer’s instruction. For each sample, 3 technical replicates were considered based on which the error bars were calculated. Polymerase chain reaction amplification was performed under the following conditions: 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute, and finally, melting at 95°C for 15 seconds and 60°C for 1 minute. The gene expressions were normalized against an internal reference gene, GAPDH, and root tissue was arbitrarily chosen to be the calibrator of tissue-specific gene expression.
Results and Discussions
CwHMGR 5′ RACE-PCR and sequencing
The functional annotation of citronella transcriptome data 39 revealed the availability of the partial sequence of HMGR gene (1344 nucleotides). The BLASTn and BLASTx results revealed the absence of around 500 nucleotides from the 5′ end of the CwHMGR coding DNA sequence. Subsequently, 5′-RACE was performed using manually designed gene-specific primers (5′-ACGGGATCTTCCCTGCCACGA-3′). The total RNA isolated from 3-month-old citronella leaf tissue was used as template. The 5′ RACE-PCR yielded a major amplification product of around 700 bp (Supplementary Figure 1). The RACE product was sequenced and aligned with the partial CwHMGR sequence. There was a clear-cut overlapping region observed in both the sequences, and full length was deduced. The final sequence was subjected to BLASTx against NR database of NCBI, which revealed that it corresponds to the adenine at position +1 of other known full-length HMGR cDNA sequences. The sequence has been deposited in NCBI GenBank (accession no. KM504513).
Multiple sequence alignment and phylogenetic analysis of CwHMGR
A BLASTP search against NR database for comparative sequence analysis of CwHMGR revealed its similarity with almost all available plant HMGR sequences. The sequences with more than 75% similarity were subjected to perform multiple sequence alignment. A total of 12 HMGR protein sequences belonging to different plant species, eg, Setaria italica, Aegilops tauschii, Brachypodium distachyon, Oryza sativa Japonica, Oryza brachyantha, Phoenix dactylifera, Arabidopsis thaliana, Oryza sativa Indica, Amomum villosum, Dendrobium huoshanense, and Dioscorea zingiberensis, were retrieved and aligned by ClustalW tool in MEGA v6.0 (Figure 1). The result showed high sequence similarity between CwHMGR and HMGR of other plants. The N-terminal end was found to be less conserved compared with the C-terminal part. This is well documented in earlier reports that, in contrast to highly conserved C-terminal catalytic domain, the N-terminal of plant HMGRs is quite diverse in both length and composition.28,41 In plants, the protein sequences of HMGR contain conserved motifs, namely, Cl-I: EMPVGYVQIP, Cl-II: TTEGCLVA, Cl-III: DAMGMNM, and Cl-IV: GTVGGGT. 41 As shown in Figure 2, all the putative HMG-CoA–binding sites (Cl-I, II, III, and IV) were found to be present in CwHMGR. Of these, motifs II (TTEGCLVA), III (DAMGMNM), and VI (GTVGGGT) were more conserved than motif I (EMPVGYVQIP). Moreover, the nicotinamide adenine dinucleotide phosphate (NADPH)–binding motifs EGC, 28 DKK, 42 GQD, 42 and C-terminal histidine (H)42,43 were also found to be conserved in CwHMGR sequence at positions E189GC, D320KK, G395QD, and H495.

Multiple alignment of amino acid sequences in CwHMGR and other plant 3-hydroxy-3-methylglutaryl-CoA reductases. The putative HMG-CoA–binding sites (Cl-I, II, III, and IV) were indicated in the shape. Alignments were performed with MEGA v6.0, and image was displayed using ESPript. Conserved residues are highlighted and shaded in boxes.

Phylogenetic analysis of 3-hydroxy-3-methylglutaryl-CoA reductase from citronella with its closest homologues using neighbour-joining method. The percentage of replicate trees in which the associated taxa are clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and are in the units of the number of amino acid differences per site. Evolutionary analyses were conducted in MEGA v6.0.
Phylogenetic analysis of citronella HMGR using neighbour-joining method in MEGA v6.0 with the other HMGRs from different plant species revealed a clear distinction into 2 large clusters representing species-specific divergence with strong bootstrap values within their nodes. The final unrooted phylogenetic tree shows dichotomy forming of 2 clusters, where HMGR sequences from monocot plants (Oryza sativa subsp. indica, Oryza sativa subsp. japonica, B distachyon, A tauschii, O brachyantha, P dactylifera, A villosum, and S italica) are grouped in cluster I, whereas cluster II included dicot species (Arabidopsis lyrata, D huoshanense, and D zingiberensis) (Figure 2). Although all HMGR sequences from monocots formed a single cluster (cluster I), CwHMGR was found to be the most closely linked with HMGR of S. italica. It was clearly observed that HMGR protein sequences from both monocot and dicot plants share high percentage of sequence similarity with CwHMGR. However, the distinct crop-specific clustering pattern could be a evidence of continuous differential evolution.
Primary sequence analysis of HMGR
The protein predicted from this cDNA contains 519 amino acid residues (GenBank ID: AIZ09174). The primary sequence analysis in ProtParam tool of ExPASy proteomic server revealed that CwHMGR has a molecular mass of 55.04 kDa with an isoelectric point of 6.57. The aliphatic index of HMGR protein is 91.76, which implies the relative volume occupied by aliphatic side chains (alanine, valine, isoleucine, and leucine). The higher aliphatic index was regarded as a positive factor for the increase in thermostability. The instability index was measured to be 48.84. As the index was more than 40, it is probably less stable in the test tube. The calculated grand average of hydropathy (GRAVY) value for CwHMGR protein sequence was 0.043. The GRAVY value is defined by the sum of hydropathy values of all amino acids divided by the protein length. The CDD search for the domain prediction revealed 4 putative domains, namely, transmembrane region (13-35 amino acids), low complexity region (64-76 amino acids), low complexity region (100-105 amino acids), and Pfam: HMGR (121-500 amino acids). To confirm the transmembrane region, we used TMHMM Web server which also predicted the presence of 1 transmembrane domain from 13th to 35th position (Supplementary Figure 2). It shows that CwHMGR belongs to class I HMGR family. Class I HMGRs consist of an N-terminal membrane domain and a C-terminal catalytic region, whereas class II enzymes lack a membrane domain. 40
Homology modelling of CwHMGR and validation
Homology modelling is usually a method of choice when a clear relationship of homology between the sequences of target protein and at least 1 known structure is found. The BLAST search against PDB database revealed 2 putative templates (PDB ID: 2Q1L and 1DQ8) of high-level identity with the target sequence, as shown in Supplementary Table 1. These templates are the crystal structures of human HMGR 2Q1L 43 and 1DQ8. 44 Due to unavailability of crystal structure of any plant HMGR protein, the homology model needs to be built based on human HMGR protein. The A-chain of 1DQ8 with a resolution of 2.05 A, due to high precision, was selected as the best template for comparative modelling. The pairwise sequence alignment of citronella HMGR and template was generated using MultiAlign, and the alignment generated was displayed in ESPript 2.2 (Figure 3).

Pairwise sequence alignment of the target protein 3-hydroxy-3-methylglutaryl-CoA reductase and A-chain of 1DQ8. The α-helices, η-helices, β-sheets, and strict β-turns are denoted as α, η, β, and TT, respectively. Similar amino acids are highlighted in boxes, and completely conserved residues are indicated by white lettering on a red background.
Based on the target-template alignment, Discovery Studio v3.5 generated 5 models of HMGR. Of these, the model with the lowest DOPE score was considered to be thermodynamically most stable and chosen for further refinement and validation. However, along with homology modelling, we also tried to predict structure for CwHMGR in I-TASSER (zhanglab.ccmb.med.umich.edu/I-TASSER/). Of the 5 predicted models, the one with best C-score was superimposed with the homology-modelled CwHMGR structure (Supplementary Figure 3). As we could not find much change in both the modelled 3D structures, the homology-based modelled structure was considered for further analysis. To assess the amount of conserved secondary structure elements, the secondary structure of the HMGR and the template was predicted and compared with the 3D structure generated. The comparison between the secondary structures of the 3D model with primary sequence–based predicted secondary structures (CONCORD) revealed that in both the cases structures were conserved and gave same configuration. The secondary structure comparison between the target and template showed a strong homology across the entire length. The identity of each α-helices and β-sheets between the target and template was presented in Table 1. The conservation of the secondary structure by MATRS proteomic server revealed the reliability of our proposed model predicted by Discovery Studio v.3.5 based on the target-template alignment.
Secondary structure comparison of HMGR and template.
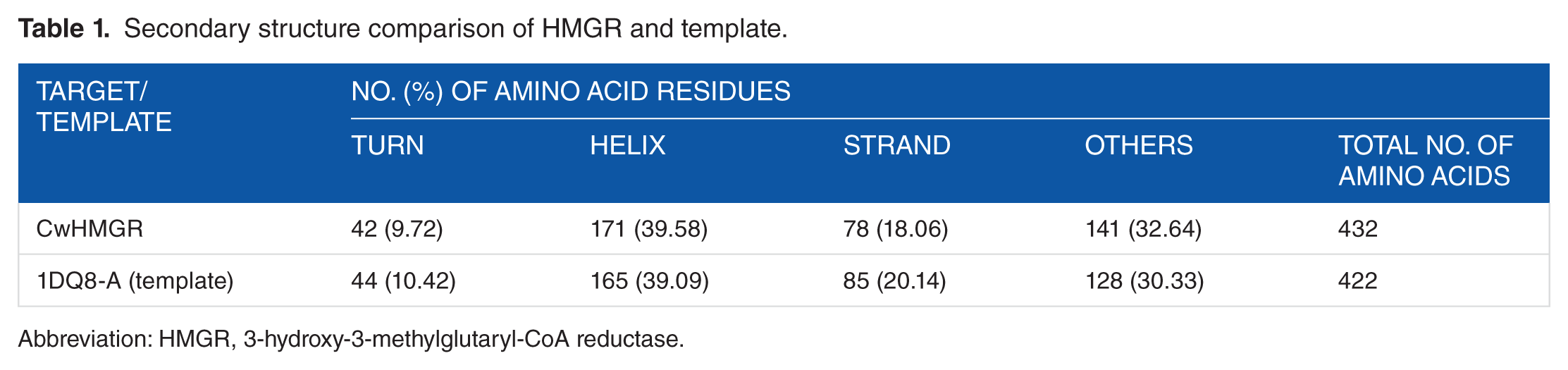
Abbreviation: HMGR, 3-hydroxy-3-methylglutaryl-CoA reductase.
PROCHECK was used to check the reliability of the backbone of torsion angles Φ, Ψ of the model, which quantifies the residue fall in the allowed regions of Ramachandran plot, as shown in Figure 4A. Ramachandran plot analysis for the modelled protein of HMGR showed that 93.0% residues fell in the most favoured regions, 6.4% residues in additional allowed regions, and 0.6% residues in the generously allowed regions, whereas no residue was reported in the disallowed regions. Compared with the template, the built 3D model had a similar Ramachandran plot which signifies the reliability of the predicted model in terms of its backbone conformation as presented in Table 2. The high quality of the structure is further evident by the G-factor of 0.03 computed in PROCHECK. The quality of our model HMGR was further supported by high ERRAT score of 94.5 (a value of ~95% shows high resolution) which indicates acceptable protein environment. 45 The VERIFY-3D results of HMGR model showed that 86.15% of the amino acids had an average 3D-1D score of greater than 0.2 indicating the reliability of the proposed model. PROVE program was used to measure the average magnitude of the volume irregularities in terms of the z score RMSD of the model. The z score RMS values of the model was 1.574 (a z score RMS value of ~1.0 indicates good resolution of structures). WHAT IF Web server analysed the coarse packing quality (−1.280), anomalous bond length (z score: 0.673), distribution of omega angles (average omega value: 176.4), and anomalous bond angles (RMS z score: 1.232) of the model protein, reflecting the good quality of the modelled protein. This further confirmed the reliability of the HMGR model.

(A) Ramachandran plot of modelled 3-hydroxy-3-methylglutaryl-CoA reductase and (B) ProSA analysis of modelled CwHMGR obtained using ProSA Web tool. NMR indicates nuclear magnetic resonance.
Comparison of Ramachandran plot statistics of HMGR model with its template 1DQ8-A.

Abbreviation: HMGR, 3-hydroxy-3-methylglutaryl-CoA reductase.
The energy profile of model and the z score value (a measure of model quality as it measures the total energy of the structures) was obtained using the ProSA program which calculated the interaction energy per residue using a distance-based pair potential. The ProSA analysis of the model HMGR (Figure 4B) achieves a z score of −8.27 and that of template was −8.60 (where the negative ProSA energy reflects reliability of the model), reflecting the good quality of the model. To check the degree of structural similarity of modelled target and template, we have measured the RMSD between equivalent Cα atom pairs along with a pairwise 3D alignment search of the template protein (1DQ8-A, human HMGR monomer structure) with the modelled structure in Discovery Studio v3.5, which showed a very low RMSD of 0.066 nm (Figure 5).The superimposition of CwHMGR with its human counterpart clearly revealed that except for a very small part in the N-terminal end, both the structures were superimposed compactly, including the catalytic domain and motifs.
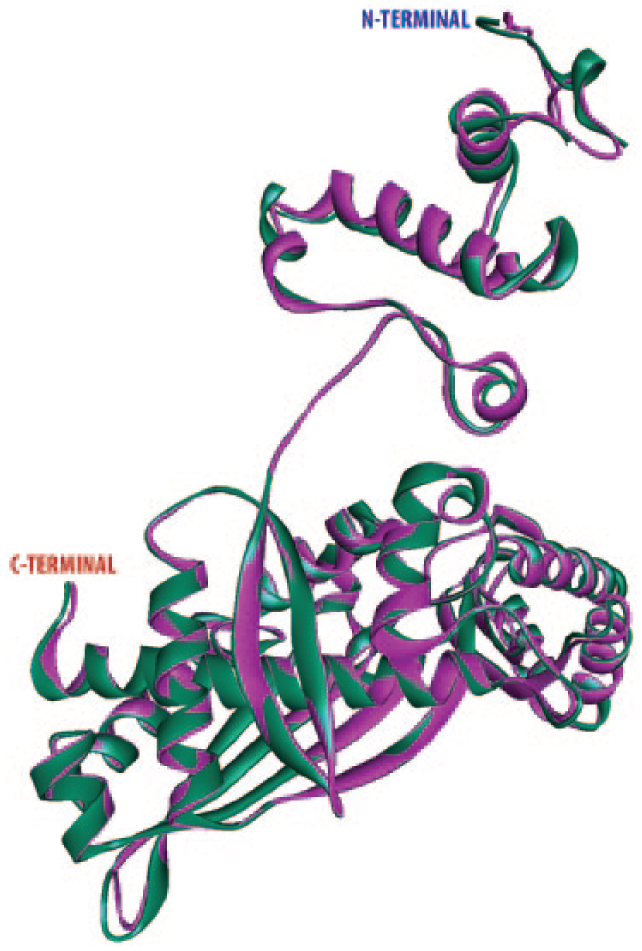
Structural superimposition of modelled HMGR (purple) and crystal structure HMGR (green) (Protein Data Bank ID: 1DQ8-A). HMGR indicates 3-hydroxy-3-methylglutaryl-CoA reductase.
Structure refinement by MD simulation
The molecular dynamic properties and stability of the predicted model of CwHMGR were examined by MD simulation for 30 ns in explicit water condition (Figure 6).The RMSD of the protein backbone atoms is plotted as a function of time to check the stability of the system throughout the simulation. The RMSD reached the overall equilibrium at 25 ns with an RMSD of ~0.08 nm after which a plateau graph was observed, as represented in Figure 6A. In the last 5 ns, it was observed that the RMSD of the system tends to be converged, indicating the system to be stable and well equilibrated. The flexibility of the model was also characterized by plotting the RMSF relative to the average structure obtained from the MD simulation trajectories. Four flexible regions have been predicted for the modelled protein structure of CwHMGR considering the RMSF value and have been represented in Figure 6B. The highest flexibility was observed in the N-terminal region. The 2 flexible regions were found to be located in N-terminal end involving the residues Val25-Arg54 and Leu55-Gly96. The N-terminal end comprises of a transmembrane domain and the linker region. The largest flexible region is the middle region of the protein involving the residues (Lys160-Gly315). This region possesses several continuous peaks with the RMSF values ranging from 0.45 to 0.25, which belong to the catalytic site directly corresponding to ligand or protein interaction. This longest flexible region possesses 4 active residues (E113, D244, K246, and G315), whereas A380 formed another peak indicating their potential binding affinity. Apart from these, some other flexible residues were also identified, namely, E113, D244, K246, G315, and A380. Based on the intrinsic dynamics, and improved relaxation of the model protein, the potential energy and total energy of the structure were calculated and the radius of gyration graph was plotted with respect to different timescales. The modelled protein remained within compact radius of gyration of 0.26 nm indicating the stable nature of the protein (Figure 6C).

MD simulation. The RMSD, RMSF, and radius of gyration graph of the modelled CwHMGR during MD simulation. (A) RMSD of backbone Cα atoms of the CwHMGR modelled structure, (B) RMSF analysis of amino acid residues of CwHMGR model structure, and (C) radius of gyration of CwHMGR modelled structure during 30 ns trajectory. All the images were generated using Xmgrace software. MD indicates molecular dynamics; RMSD, root mean square deviation; RMSF, root mean square fluctuation.
Detailed structural study of HMGR
CwHMGR of 519-amino-acid-long protein was found to depict extensive homology with other plant HMGRs and consists of 1 transmembrane domain and 1 catalytic domain. The predicted 3D model of CwHMGR had a typical spatial structure of HMGRs. It consists of 22 α-helices and 9 β-strands distributed across 3 domains. The structure has a small N domain (1-12 amino acid residues), transmembrane domains (13-76 amino acid residues), and large C domain (121-422 amino acid residues). The membrane domains of plant HMGRs (class I) contained 2 membrane spanning helices. 44 In the predicted model, only 1 distinct transmembrane region has been observed comprising 13-35 number residues consisting of 3 α-helices. The active site of modelled HMGR consists of 2 pockets: NADPH-binding pocket and the substrate-binding pocket. Comparison of modelled CwHMGR protein with human HMGR revealed that the catalytic domain (C domain) of CwHMGR consisted of 3 domains, including the small helical N-terminal domain, the large L domain containing 2 HMG-CoA–binding motifs and an NADP(H)-binding motif, and the smallest S domain harbouring an NADP(H)-binding motif (Figure 7). The 3D structure of CwHMGR strongly resembled human HMGR, 44 indicating that they had potential catalytic similarities.
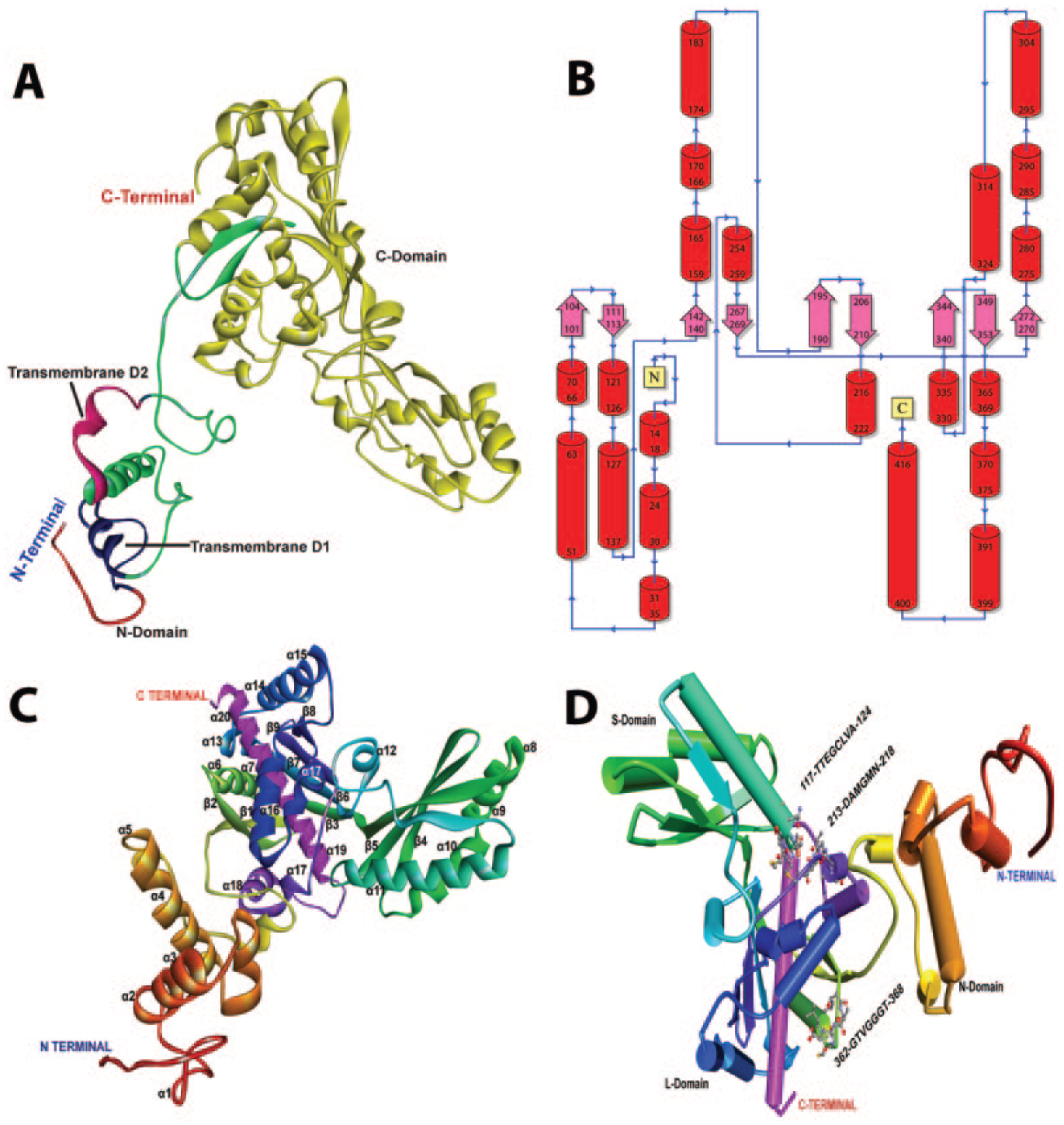
Detailed structure of CwHMGR. (A) Three-dimensional (3D) structure of CwHMGR indicating the 3 domains, (B) the topology of CwHMGR, (C) the detailed 3D structure of CwHMGR indicating α-helices and β-sheets, and (D) modelled CwHMGR indicating the catalytic motifs.
Tissue-specific differential expression
The RT and qRT-PCR results indicate that HMGR shows almost equal level of expression in both leaf sheath and root tissue. The enzyme HMGR, being a cytosolic enzyme, is the first rate-limiting enzyme of MVA pathway and is responsible for the production of a number of diverse products such as squalene, ubiquinones, sterols, and terpenes, 47 which may be produced in both underground and above-ground parts of this plant (roots and shoots). Therefore, the expression of HMGR may be equal in both root and leaf sheath tissue, but the flux of intermediary product may be diverted to different pathways in different tissues to produce varying metabolites (Figure 8). This might be the reason for low expression in leaf compared with leaf sheath and root.

Tissue-specific expression profiling of CwHMGR in 3 different tissues (root, leaf, and sheath). (A) Reverse transcription-polymerase chain reaction result and (B) quantitative real-time polymerase chain reaction result.
Citronella is a highly useful medicinal aromatic plant, and so increasing its level of essential oil synthesis is of great interest to commercial producers. Thus, the upregulation of the key enzyme of MVA pathway, ie, HMGR, will be a significant step for increased essential oil production. This study is the first ever report on characterization of complete coding region of HMGR in C winterianus. In the modelled structure, the presence of single transmembrane domain categorized the protein into class I HMGR family. The 4 most conserved catalytically important domains are positioned in the CwHMGR 3D structure in such a way that it forms a catalytic pocket. Along with this, we also identified the conserved catalytic motifs, which could be crucial targets for any kind of modification in CwHMGR protein. This study is based on in silico methods, and so, combinatorial methods involving biochemical, enzyme kinetics, and site-directed mutagenesis will be needed to confirm these predictions. However, this is a promising first step towards opening up better avenues for metabolic engineering of secondary metabolite pathways.
Footnotes
Acknowledgements
The authors are highly grateful to Dr. Iswar Boruah for providing plant materials for the experiment. They would like to thank the Distributed Information Centre, Department of Agricultural Biotechnology, AAU, Jorhat, for providing computational facility. They deeply acknowledge the Department of Science and Technology, Government of India, for Inspire Fellowship (IF120251) to the first author (KD).
Peer review:
Four peer reviewers contributed to the peer review report. Reviewers’ reports totalled 1005 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: They deeply acknowledge the Department of Science and Technology, Government of India, for Inspire Fellowship (IF120251) to the first author (KD).
Declaration of conflicting interest:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
PS conceived and designed the experiments. KD and LP analysed the data, wrote the first draft of the manuscript, and jointly developed the structure and arguments for the paper. KD performed the wet lab analysis. PS and MKM contributed to the writing of the manuscript and agree with manuscript results and conclusions. MKM made critical revisions and approved final version. All authors reviewed and approved the final manuscript.
Disclosures and Ethics
As a requirement of publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including, but not limited to, the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.