
Abstract
Synapses are the site for brain communication where information is transmitted between neurons and stored for memory formation. Synaptic degeneration is a global and early pathogenic event in neurodegenerative disorders with reduced levels of pre- and postsynaptic proteins being recognized as a core feature of Alzheimer’s disease (AD) pathophysiology. Together with AD, other neurodegenerative and neurodevelopmental disorders show altered synaptic homeostasis as an important pathogenic event, and due to that, they are commonly referred to as synaptopathies. The exact mechanisms of synapse dysfunction in the different diseases are not well understood and their study would help understanding the pathogenic role of synaptic degeneration, as well as differences and commonalities among them and highlight candidate synaptic biomarkers for specific disorders. The assessment of synaptic proteins in cerebrospinal fluid (CSF), which can reflect synaptic dysfunction in patients with cognitive disorders, is a keen area of interest. Substantial research efforts are now directed toward the investigation of CSF synaptic pathology to improve the diagnosis of neurodegenerative disorders at an early stage as well as to monitor clinical progression. In this review, we will first summarize the pathological events that lead to synapse loss and then discuss the available data on established (eg, neurogranin, SNAP-25, synaptotagmin-1, GAP-43, and α-syn) and emerging (eg, synaptic vesicle glycoprotein 2A and neuronal pentraxins) CSF biomarkers for synapse dysfunction, while highlighting possible utilities, disease specificity, and technical challenges for their detection.
Introduction
The central nervous system (CNS) can be subject to numerous pathological conditions, which can affect its development, functionality, or cause premature cell death, resulting in neurodevelopmental, neuropsychiatric, and neurodegenerative disorders. Although these disorders have different etiologies and pathophysiological mechanisms, many of them have some degree of dysfunction and alteration of the synapses and can thus be categorized as synaptopathies.1,2 In this review, we will discuss how synapses are affected in the most common diseases affecting the CNS, and how advances in synaptic biomarker discovery provide new tools for the study of those diseases. We will mainly focus on neurodegenerative conditions, and in particular on Alzheimer’s disease (AD), which is the predominant cause of dementia affecting approximately 50 million people worldwide. 3 Although the exact mechanisms of synaptic loss and dysfunction in the different diseases are still poorly understood, there is evidence that a reduction in synaptic activity and density is one of the earliest events in many of the diseases of the CNS and may even appear before neuronal loss.4,5 The significant role of synapse dysfunction in the disease pathology and progression of synaptopathies has therefore prompted a keen interest in detecting and quantifying synaptic proteins. Molecular brain imaging 6 and analysis of cerebrospinal fluid (CSF) 7 are used in conjunction to study synaptic proteins, with the aim of using them as biomarkers for prognosis, to follow disease progression and to evaluate effects of drug testing.
Pathophysiology of Synaptic Dysfunction and Loss
Synaptic functions
The neuronal synapses are the functional units of neurotransmission in the brain, with an estimated 100 trillion interconnecting synapses 8 in an elaborate and complex network. Synapses are formed during development and the early postnatal period. After reaching the maximum density at 2 to 4 years of age,9,10 in the following years synapses are physiologically eliminated in a process known as pruning. 11 Synapses that survive to adulthood are the ones stably maintained, although we have a certain degree of synapse formation and elimination throughout life. 12
Neuronal signal transmission in the CNS requires the presence of functional synapses, with properly arranged pre- and postsynaptic compartments. The presynaptic compartment contains all the structures for formation, storage, and release of neurotransmitter-containing vesicles. Following an action potential, the increase of Ca2+ in the presynaptic terminal triggers synaptic vesicles to fuse with the presynaptic membrane upon which neurotransmitters are released into the synaptic cleft. 13 Subsequently, neurotransmitters interact with receptors on the postsynaptic compartment (the dendritic spine), and through the activation of different signaling pathways 14 the neuronal signal is transmitted further. Synapses can be excitatory or inhibitory, using glutamate and GABA, as neurotransmitters, respectively. 15 The dendritic spines are the primary location of excitatory synapses.
Synapse formation, maturation, and elimination is a dynamic series of events that can be defined as synaptic plasticity. Processes representing synaptic plasticity are phenomena termed long-term potentiation (LTP) and long-term depression (LTD), through which, during memory formation, signaling via preferred synapses is enhanced or reduced. Selection of synapses seems to be activity-dependent, LTP is usually considered as a protective mechanism and LTD as inductive of elimination. 16 These 2 processes are considered the basis for memory formation and storage.17,18 LTP is identified by the addition of new receptors at the postsynaptic density (PSD) and the consequent enlargement of the spine head resulting in transmission of a stronger signal. 19 On the contrary, during LTD a series of events lead to spine shrinkage and elimination. 18 Many different mechanisms for synaptic elimination have been suggested (for extensive review, see Cardozo et al 20 and Maiti et al 21 ) Elimination of weaker and unnecessary synapses and maintenance of the stronger ones are processes that balance each other, to ensure proper connectivity between brain regions and signal refinement.22,23 For proper synaptic activity, a balance is needed, and alterations between synapse formation and elimination can cause synaptic dysfunction and impaired brain network activities. 21 To understand pathological mechanisms and at which stage synapses are affected is of utmost importance to define targets and intervention strategies.
Synapse and neuronal loss in brain disorders
As mentioned, a balance in synapse formation and pruning is essential for proper connectivity and brain functionality. For instance, excessive synaptic pruning during adolescence is one of the hypothesized mechanisms for schizophrenia, which most commonly manifests with an onset in late adolescence or early-adulthood.24-26 The term “synaptopathy” is applied to refer to all diseases that are characterized by a progressive synaptic dysfunction and loss. 20 AD, the most common neurodegenerative disease, can be therefore considered both a synaptopathy and a proteinopathy.
AD pathology is identified by the presence of extracellular deposits of amyloid-β (Aβ) plaques, formed by the aggregation of Aβ peptides, and neurofibrillary tangles (NFTs) that are intraneuronal accumulations of hyperphosphorylated and truncated tau protein, respectively.27,28 Along with these main hallmarks, gliosis, neuroinflammation,29-31 and vascular dysfunction32,33 are also present, which reflects the complexity of AD. However, it is synaptic loss which best correlates with cognitive symptoms34-36 and it is also apparent in the early stages of the disease pathophysiology.37,38 The number of synapses in the brain decreases during normal aging but this decrease is exacerbated in AD and, consequently, the synapse-to-neuron ratio is lower in AD brains compared with age-matched non-demented individuals. 39 In AD, brain biopsies show synaptic loss in neocortical regions and the hippocampus,40,41 the latter showing the greatest reduction by approximately 50%.42-44
How the major AD hallmarks, tau and Aβ, pathologically interact with synapses needs more investigation. However, most studies identified the oligomeric forms of Aβ and tau, rather than larger aggregates, to be the synaptotoxic species.45-49 Both in vivo
50
and ex vivo
51
Aβ oligomers (Aβo) disrupt LTP, probably interfering with NMDAR (N-methyl-
Moreover, as points of transmission of signals between neurons, synapses seem also to help the spreading of the pathology via prion-like mechanisms, and some studies show the possibility of Aβo and tau oligomers to be transferred from neuron to neuron.64-69
Tau aggregation, without Aβ pathology, is also a pathological hallmark of other neurodegenerative diseases, the so-called tauopathies. 70 Tauopathies include, among others, some forms of frontotemporal lobar degeneration (FTLD), namely FTLD-tau, progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD). Although all tauopathies have in common the presence of tau aggregates in the CNS, the characteristics of these aggregates differ among them and are different from the NFTs of AD. PSP shows filamentous aggregates in astrocytes and oligodendrocytes, while in CBD tau accumulation in neurons is less fibrillar and in astrocytes it accumulates in the form of astrocytic plaques. 71 As introduced above, oligomeric tau has been connected to synaptic damage through different pathways, 63 also involving activation of microglia and astrocytes through inflammatory processes, 72 and animal models of tau pathology show early synaptic loss prior to neuronal death.73,74
As the combination of Aβ and tau pathology define AD, accumulation of aggregated α-synuclein (α-syn) is the pathological feature of several diseases, which are commonly collectively referred to as α-synucleinopathies. 75 Among the most common synucleopathies are Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), dementia with Lewy body (DLB), and multiple system atrophy (MSA). PD, PDD, and DLB show accumulation of the so-called Lewy neurites (LN) and Lewy bodies (LB), where α-syn is the principal component. 76 The physiological role of α-syn at the synapse is not precisely understood yet 77 ; however, it is commonly accepted that its dyshomeostasis and accumulation leads to cell damage and it is responsible for synaptic impairment and neuronal damage.78,79 Alpha-syn, localized mainly presynaptically, is involved in synaptic vesicle regulation and trafficking,80,81 and in the SNARE complex formation.82,83 It also interacts with membrane lipids and can associate with mitochondria, 84 the Golgi-endoplasmic reticulum system, 85 and the endolysosomal system. Pathologic α-syn can interfere with all these organelles, consequently impairing-related pathways.86,87 The protein can change its conformation and aggregate, giving rise to oligomers, fibrils, and larger aggregates. 88 Which form is responsible for toxicity is still a matter of debate. However, as discussed for AD, the oligomeric form of α-syn has been suggested to be the responsible for the synaptic damage in dopaminergic neurons,89,90 and the possibility for oligomeric α-syn of spreading in a prion-like manner has been proposed.91,92 At a cellular level, typically PD and DLB are distinguished from MSA, inasmuch the accumulations of LN and LB are mainly present in neurons, while in MSA, α-syn accumulation appears in oligodendrocytes. 93 Moreover, MSA inclusions seem to be more compact and aggressive, 94 in line with the increased severity of the disease. 95 However, it was most recently reported that also neurons in MSA show α-syn oligomers depositions. 96 A recent study showed that α-syn filaments differ in DLB and MSA. 97 PD and DLB are usually distinguished based on the symptoms, with DLB being the second most common type of dementia. 98 PD, similar to AD, starts many years before symptoms become overt and at that point, patients had already lost up to 60% motor neurons in the substantia nigra. 99
Nonetheless, α-syn was first identified and characterized in relation to AD when it was found to be a major non-amyloid beta component of Aβ plaques. 100 In fact, Lewy pathology can be also found in over half of all patients with AD.101-103 To further complicate the clinical picture, α-syn depositions are found in tauopathies like PSP and CBD, and NFTs have been found in PD brains.104,105
Comorbidities and co-occurrence of different pathologies make diagnosis of these diseases challenging. Synapse damage is a common and early first change in the disease development and prolonged synaptic damage can lead to synaptic loss. Neuronal damage and death seem to be a follow-up event seen only at later stages. For these reasons, the investigation of synaptic biomarkers has the potential to find a way to diagnose the disease in its early stages and also to give us information on the main pathological mechanisms involved.
Current Climate of Fluid Biomarkers in Dementia
Imaging and CSF biomarkers
The core CSF biomarkers for AD (Aβ42/Aβ40, total-tau, and phospho-tau), reflecting the defining Aβ and tau neuropathologies, consistently demonstrate diagnostically significant changes across studies 106 and now have prominent positions in biological and diagnostic criteria for AD.28,107 The concentrations of these core AD biomarkers, however, are no different from healthy controls in the majority of dementias outside of the AD continuum108,109 which can be of great utility in the differential diagnosis of patients with cognitive symptoms. An exception can be made for Creutzfeldt–Jakob disease (CJD), which presents vastly increased levels of t-tau, whereas the concentrations of p-tau181 remain normal or only marginally changed in CJD.110,111
Together with CSF biomarkers, positron emission tomography (PET) and magnetic resonance imaging (MRI) are used to provide a clearer view of pathology and atrophy patterns in the brains of living humans. MRI allows for the measurement of brain atrophy and provides information on regional, structural, and functional integrity of the brain. 112 In the research of neurodegenerative disorders, PET tracers for protein aggregation such as Aβ113,114 and tau, 115 as well as glucose metabolism as a measure for neuronal activity116,117 and synaptic density, 118 have been developed. Together with CSF biomarkers, MRI and PET are nowadays included in the research diagnostic criteria for AD.28,119 However, the availability of PET scans is limited and when possible expensive, thus it is not always applicable.
Blood biomarkers
In certain instances, the biomarkers field is rapidly evolving from CSF into blood, which is a more easily accessible biological fluid. Despite the latest advancement in developing CSF biomarkers for synaptic integrity and large high-resolution mass spectrometry proteomic studies demonstrating the presence of synaptic proteins in blood,120,121 to date no studies have shown positive results for any pre- or postsynaptic biomarkers in blood correlating to any neurodegenerative disease phenotype.
The advancement of ultrasensitive methodologies has enabled, however, the detection of the CSF core biomarkers and neuronal injury, like neurofilaments, in blood. New evidence from high-resolution mass spectrometry,122,123 single molecule array (Simoa), 124 and fully automated immunoassays, 125 which are highly sensitive and alleviate confounding matrix effects in blood, suggests that Aβ peptide ratios are specific markers of individuals with Aβ- positive brain scans. In addition, recent evidence has shown that plasma p-tau181 concentrations are higher in individuals with AD dementia than in healthy controls. 126 Plasma p-tau181 correlates with tau PET in Aβ-positive AD individuals and, encouragingly, can accurately identify elderly controls and mild cognitive impairment (MCI) individuals with a positive Aβ-PET scan (area under the curve [AUC] > 0.85).127-129 Conversely, although significant increase of plasma t-tau has been vastly observed in individuals with AD, the plasma t-tau levels between control, MCI, and AD groups substantially overlapped. 130
The neurofilaments are cytoskeletal protein abundantly expressed in neuronal axons, among which neurofilament light polypeptide (NfL) is the smallest of the neurofilament proteins (for a detailed review on neurofilament structure and function, please see Khalil et al 131 ). A moderate-to-good correlation between NfL concentration in blood and CSF has been observed in several studies and many CSF findings of increased NfL in neurodegenerative diseases have subsequently been replicated in blood. 132 Although not a specific marker for AD, blood NfL has the potential to track or predict many aspects of neurodegeneration, including cognitive performance, 133 the degree of postmortem pathology, 134 structural imaging, 135 and glucose metabolism.136,137
Fluid Biomarkers for Synapse Pathology
The pathophysiology of synaptopathies and the significance of synapses in cognition make a convincing argument for the need and use of biomarkers of synapse pathology as representation of cognitive and synaptic function. Clinically, synaptic biomarkers may link synaptic degeneration with the cognitive status and decline of the patient, and they could be implemented together with cognitive tests to have a more precise description of the patient’s symptoms, especially at early stages. Moreover, synaptic biomarkers can help to understand the underlying pathological processes ongoing during cognitive diseases, as different proteins could reflect different mechanisms, thus helping the diagnosis. In addition, synaptic biomarkers can also be used during drug development, to monitor the efficacy of treatments on synaptic functioning in drug trials.
Pre- and postsynaptic biomarkers
Biomarkers for synaptic dysfunction can be divided into pre- and postsynaptic biomarkers depending on the localization of the protein. The presence of synaptic proteins in CSF was first demonstrated in the late 1990s,138,139 but for a long time, most studies still involved postmortem brain tissue. However, in the last decade, advances in mass spectrometry and immunoassays have allowed the accurate quantification of synaptic proteins in biofluids. As of today, there are 4 main presynaptic biomarkers, growth-associated protein 43 (GAP-43), synaptosomal-associated protein 25 (SNAP-25), synaptotagmin-1, and α-syn, and 1 postsynaptic marker, neurogranin.
GAP-43
GAP-43 is a presynaptic protein which plays an important role in memory and information storage. 140 It is anchored on the cytoplasmic side of the presynaptic plasma membrane and is mainly expressed in the hippocampus, entorhinal cortex, and neocortex of the adult brain. At the synapse, upon intracellular Ca2+ increase, GAP-43 is phosphorylated by protein kinase C. This leads GAP-43 to interact, among others, with synaptophysin and SNAP-25, facilitating synaptic vesicle recycling. 141 Studies have found GAP-43 CSF levels to be significantly increased in patients with AD compared with controls 142 and also other neurodegenerative disorders. 143 CSF GAP-43 levels were also increased in preclinical and clinical patients with AD compared with controls. However, in an antibody-based explorative study, no significant changes in patients with PD or DLB were found in comparison with controls. 144 Altered CSF GAP-43 levels have also been reported in progressive multiple sclerosis (MS),143,145 inflammation, 146 stroke, 147 and PD, 109 but not in frontotemporal dementia (FTD). 109
SNAP-25
SNAP-25 is a presynaptic protein with a key role in neuronal survival and cognitive function due to its essential part in vesicular exocytosis, neurite outgrowth, and LTP. 148 SNAP-25, together with vesicle-associated membrane proteins (VAMPs) and syntaxins, forms SNARE complexes, which mediate synaptic vesicle apposition to the presynaptic membrane thus allowing for the Ca2+-triggered vesicle fusion during exocytosis. 149 SNAP-25 has, in various studies using both enzyme-linked immunosorbent assay (ELISA) and mass spectrometry-based assays, shown to have significantly higher CSF levels in AD, even at a very early stages.7,150-152 Increased CSF levels of SNAP-25 have also been found in patients with PD 153 and patients with sporadic CJD. 154 In addition, SNAP-25 has been associated with several psychiatric diseases such as attention deficiency hyperactivity disorder (ADHD), schizophrenia, and bipolar disorder. 149 Furthermore, there are 2 splicing variants of SNAP-25: SNAP-25A and SNAP-25B. Mass spectrometry–based methods to quantify both total SNAP-25 and the 2 isoforms have therefore been developed to study potential differences in the roles of the isoforms of SNAP-25 in disease. Nine amino acids differentiate the 2 protein isoforms, which also differ in their effects on neurotransmission. 155 To our knowledge, no studies have investigated the different isoforms in CSF. However, a postmortem brain tissue study by Barakauskas et al 155 found significantly decreased levels of total SNAP-25 and SNAP-25A but not of SNAP-25B, indicating a specific differential expression of SNAP-25A in schizophrenia.
Synaptotagmin-1
Synaptotagmin-1 is a calcium sensor vesicle protein vital for fast synchronous neurotransmitter release in hippocampal neurons. 156 It is a transmembrane protein anchored in the vesicle membranes containing 2 Ca2+-binding domains. In response to Ca2+-binding at elevated concentrations, synaptotagmin-1 triggers the vesicle fusion, but the exact molecular mechanisms remain to be elucidated (for review see Park and Ryu 157 ). Initial CSF studies of synaptotagmin-1 found it to be decreased in a CSF pool from patients with early-onset AD compared with a CSF pool from healthy controls. 138 Two decades later, Öhrfelt et al 158 quantified synaptotagmin-1 in individual CSF samples from patients with AD, MCI, and controls, demonstrating significantly increased concentrations of synaptotagmin-1 in patients with AD and MCI, the highest being MCI due to AD. These findings have been corroborated in a recent study where synaptotagmin-1 was quantified in patients in the AD continuum and cognitive decline from other dementias.159,160
In the same study by Tible et al, in addition to synaptotagmin-1, the concentrations of GAP-43 and SNAP-25 were quantified. All 3 presynaptic proteins were significantly increased in AD and MCI-AD compared with the other disorders. However, only SNAP-25 and GAP-43 levels were also significantly higher in AD versus MCI-AD. Only synaptotagmin-1 concentrations were significantly lower in other neurodegenerative disorders compared with controls. Recently, Clarke et al 161 compared synaptotagmin-1 and SNAP-25 concentrations in patients with FTD and demonstrated increased levels in patients with AD biomarker profile compared with those patients with an FTD profile.
Alpha-synuclein and its forms
Alpha-syn is a key player in the etiology of different neurodegenerative conditions and as such, it has been studied as a possible biomarker for their detection. However, besides being a possible cause for diseases, it is also a presynaptic protein, taking part in many synaptic processes as previously described, which is why it is important to include it in this review. The synucleins family comprises α-, β-, and γ-synuclein, which are soluble proteins encoded by 3 different genes. Among them, α-syn is the most studied. 162
Total α-syn
Studies of α-syn in CSF have mainly been focused on α-synucleinopathies and were based on immunological assays measuring total α-syn (t-α-syn); however, they have been largely inconsistent. For instance, in PD compared with controls, t-α-syn has been found in several studies to be slightly decreased163-165 which is supported by several meta-analyses which concluded that there are significantly lower levels of t-α-syn in PD (10%-15%). However, in other studies no significant difference has been found,166,167 and the diagnostic performance of t-α-syn is not considered sufficient for clinical utility due to significant overlap between the populations.168-170 Other synucleinopathies, like DLB and MSA, have also shown a similar decrease compared with healthy controls, while tauopathies such as PSP and CBD seem to show no significant difference.163,165,171 For AD in comparison with healthy controls, t-α-syn levels seem to be elevated171-174; however, several studies showed no significant difference.175-179 Patients with CJD, on the other hand, have a more pronounced increase in CSF t-α-syn, both compared with controls and with other neurodegenerative diseases.178,180,181 An explanation for the inconclusive findings of CSF α-syn is that leakage into the CSF from synapse breakdown occurs simultaneously as α-syn is retained in pathological inclusions. In addition, the extensive reduction in synapse number over time might lead to a decrease of α-syn production. Together these events might contribute to the confounding results.166,182 Another contributing factor for the varying results might be due to technical variation such as handling of samples or quantification methods leading to low reproducibility. 182 Moreover, α-syn is largely expressed outside of the CNS and highly abundant in blood, with red blood cells (RBCs) as its major source. Thus, blood contamination during CSF acquisition might represent another source of variation, skewing the t-α-syn concentration results in CSF.183,184
Despite the possible problems just discussed, there have been many studies evaluating α-syn as blood biomarker for dementias. Studies for α-syn in plasma and serum in PD have all shown similar conflicting results as in CSF.185-188 However, a meta-analysis indicates that plasma t-α-syn is significantly higher in PD than in controls. 189 In a study by Laske et al, 190 decreased serum concentrations of t-α-syn in DLB were found but with no difference for AD compared with controls. There are also a few studies on RBC t-α-syn191-193 which showed significantly decreased levels of the protein in PD and AD compared with controls. Studies on t-α-syn in saliva have also been performed, but with limited success in differentiating PD from controls.194-196
Oligomeric, phosphorylated, and aggregated forms of α-synuclein
The inconclusive results of t-α-syn as a diagnostic biomarker have sparked research into pathological forms of α-syn, such as oligomeric (o-α-syn), phosphorylated (Ser129) (p-α-syn), and aggregated forms of α-synuclein. Oligomeric α-syn in CSF seems to be increased in PD compared with controls 169 but not in AD and DLB.174,197 Furthermore, Parnetti et al 198 found that the diagnostic accuracy of PD can be improved by using the ratio of oligomeric/total α-syn in CSF. In plasma, 199 serum, 200 and RBC,193,201 significantly elevated levels have been reported for PD, but also non-significant results exist.202-204 In a study by Vivacqua et al, 205 increased saliva levels of o-α-syn were found for PD. Phosphorylated α-syn, one of the main disease-associated posttranslational modifications (PTMs), 206 is hard to quantify due to its low CSF concentration, but similar to the oligomeric and the total form, it has been found elevated in PD 169 and its diagnostic accuracy increases when its ratio to other α-syn forms are used. 207 Phosphorylated-α-syn has also been indicated to be elevated in CJD 181 and not increased in AD.174,179 Plasma p-α-syn has been found to be significantly increased in PD compared with controls.185,203 For the measurement of pathogenic α-syn aggregates in CSF, aggregation assays have been developed. Assays based on real-time quaking-induced conversion (RT-QuIC) or protein misfolding cyclic amplification (PMCA) have shown very promising results (specificity > 95%, sensitivity > 80%) in discriminating synucleinopathies (PD, MSA, and DLB) from nonsynucleinopathies (AD and controls).208-210
Neurogranin
Neurogranin is an intracellular 7.5-kDa protein, concentrated in the dendritic and postsynaptic compartment of synaptic spines of neurons.211,212 There it binds via its central IQ domain 213 to the Ca2+-signaling mediator calmodulin, enhancing signaling for processes important in memory formation and to phosphatidic acid at the inner plasma membrane. 214 A neurogranin knockout mouse model showed deficits in spatial memory and synaptic plasticity. 215 In a first study, CSF neurogranin was shown by immunoprecipitation and Western blot to be increased in AD. 216 After the development of immunoassay methods using ELISA, 217 Singulex, 218 and Mesoscale, 219 these findings have been verified in several studies and neurogranin consistently showed increased levels in CSF of AD patients as compared with controls.121,220-224 This increase appears to be specific for AD, as CSF from patients with other neurodegenerative diseases, with the exception of CJD, 225 do not show such an increase.161,226,227 High levels of neurogranin in CSF during prodromal AD have been shown to be predictive of more rapid progression toward AD.217,219
Besides full-length neurogranin, CSF contains mainly fragments of the C-terminal half (with a variety of different truncations at their C-terminal and N-terminal ends).216,217 Two intracellular enzymes have been identified that can generate cleavages in the functionally important IQ domain and at the very C-terminal end (calpain-1 and prolyl endopeptidase, respectively). 228 Whether these different fragments of neurogranin have roles in different physiological or pathophysiological functions is still unknown. In a comparison study, different ELISAs and the Singulex assay were found to have similar performance in predicting AD, in spite targeting different parts of neurogranin. 229 However, this does not rule out that particular neurogranin fragments could yield more discriminatory power to detect AD. Overall, it can be said that neurogranin may be a useful biomarker in CSF to detect early degeneration of neurons and it appears to be fairly specific for AD among several tauopathies.
Plasma concentrations of neurogranin are detectable with conventional ELISAs but are unchanged in AD and do not correlate with CSF neurogranin, probably due to the contribution of peripherally expressed neurogranin peptides to blood neurogranin measurements.121,220
Emerging synaptic biomarkers
Recently, other studies identified more synaptic proteins in CSF, which have been investigated without success so far or that show promise as synaptic biomarkers, thus worth to be mentioned in this section. Wesenhagen et al have recently reviewed 29 proteomic studies that investigated AD-related changes in CSF protein abundances. In total, 97 proteins, including the synaptic proteins neurofascin, NPTX1, NPTX2, and neurexin 1, were reported by 2 or more studies and associated with AD. 230 One of the reviewed studies 231 reported a synaptic biomarker panel where only the 3 synaptic proteins neurofascin, NPTX1, and neurexin 1 were significantly lowered in AD. Similarly, Lleó et al 232 found that 6 synaptic proteins, calsyntenin-1, glutamate receptor 4 (GRIA4), neurexin-2A, neurexin-3A, syntaxin-1B, and thy-1 membrane glycoprotein, were increased in CSF in preclinical AD even before the core CSF biomarkers for neurodegeneration.
In explorative proteomics, high-resolution separation methods such as gel electrophoresis, isoelectric focusing, and high-performance liquid chromatography are used in conjunction with mass spectrometry and bioinformatics to study differences in protein expression due to diseases, genetic variations, or therapy. A major advantage of using an explorative approach to study protein abundances is that many hundred proteins and protein variants can be studied simultaneously without existing hypotheses or bias. Thus, the discovery of novel biomarkers could lead to new insights on disease mechanisms and eventually the formulation of novel hypotheses. However, using the explorative approach to identify biomarkers in biofluids from individual patient samples is challenging and the overlap of identified biomarker candidates among these studies has historically been relatively low. These discrepancies may be due to a low number of study participants, differences in sample handling, and other analytical parameters. Another possible approach to identify new candidate biomarkers is setting up targeted assays based on proteins of interest from studying the literature and/or public databases. Commonly shotgun proteomics, to identify possible proteins of interest, is also used in the selective process. In this way, several potential biomarker candidates can be validated in a targeted setting in larger cohorts. Among emerging synaptic biomarkers, of special note are neuronal pentraxins and the synaptic vesicle glycoprotein 2A.
Neuronal pentraxins
Neuronal pentraxin I (NPTX1, also called NP1) and II (NPTX2, also called NP2), and the neuronal pentraxin receptor (NPTXR) are widely expressed at excitatory synapses, where they bind to AMPA receptors and are suggested to be involved in synaptic plasticity.233,234 All 3 neuronal pentraxins have lately received much attention and have been shown in several studies to have decreased CSF levels in AD and MCI groups compared with controls.231,235-242 CSF pentraxin levels also correlate with cognitive performance and hippocampal volume.150,242,243 Few studies have been performed on other diseases but NPTXR has also been associated with other neurological diseases such as MS 244 and FTD. 245 Furthermore, in a study by Magdalinou et al 246 both NPTXR and NPTX1 were found to be decreased in between atypical parkinsonian disorders (PSP, MSA, CBD) and controls.
SV2A
Recent studies using [11C]UCB-J PET have identified SV2A as the first in vivo marker of synaptic density 247 which demonstrates widespread synaptic loss in AD. 118 SV2A is a synaptic vesicle transmembrane protein, which in brain is widely expressed in neurons. 248 SV2A has been described to be located in the dense-core vesicles249,250 and in small synaptic vesicles, 251 most probably in both. Although its exact mechanism needs more investigation, it is involved in regulation of neurotransmitter release252,253 and expression and trafficking of synaptotagmin. 254 Compared with the typical pattern of hypometabolism seen in AD using [ 18 F]FDG, the spatial extent of decreases in [11C]UCB-J uptake was significantly more confined. The reduction in hippocampal binding is in line with the early loss of entorhinal cortical cell projections to the hippocampus, and reductions of hippocampal SV2A seen in postmortem studies in AD brain tissue.255,256 More recently, changes in [11C]UCB-J PET have been observed in PD, 257 PSP, 258 cortical basal syndrome, and epilepsy 247 suggesting that SV2A could be a global marker for synaptic density, unlike CSF synaptotagmin-1, SNAP-25, GAP-43, and neurogranin, which are rather specific to AD or amyloidopathies. Recently, SV2A has been detected in CSF and shown to be reduced in AD 259 ; however it is yet to be determined whether CSF SV2A can be used as a marker for synaptic density in other dementias and whether it has a meaningful correlation with [11C]UCB-J (Figure 1).

Synaptic and neuronal biomarkers location. The picture is a schematic representation of the most studied synaptic biomarkers described in this review. As it can be noticed, most of the candidate biomarkers are localized presynaptically, with the exception of neurogranin and neuronal pentraxins (NPTX), which has also been described to be present presynaptically. 260 Many proteins are involved in synaptic vesicle assembly and neurotransmitters release, like synaptotagmin-1 (syt 1), synaptophysin, SNAP-25, and SV2A. 248 α-Synuclein (α-syn) can be found as a soluble form in the cytoplasm, but also associating with membrane lipids as, for instance, with synaptic vesicles and mitochondria. 87 GAP-43 shows high density in the presynaptic terminal, where depending on its phosphorylation status, participates in neuronal growth modulating actin or in synaptic plasticity modulating synaptic vesicle trafficking. 141 Together with actin filaments and microtubules, neurofilaments are cytoskeletal elements of the neurons, providing mechanical strength and stability. 131 Tau protein, mainly expressed in axons, binds to tubulin and induce its polymerization into microtubules, which support axon outgrowth and elongation. 261 α-syn indicates synuclein; DCV, dense-core vesicles; GAP-43, growth-associated protein 43; LTP, long-term potentiation; NPTX, neuronal pentraxin; SNAP-25, synaptosomal-associated protein 25.
Miscellaneous: other emerging synaptic biomarkers
The Rab family are key synaptic proteins involved in both recycling of neurotransmitter receptors and exocytosis of neurotransmitters. Of special note is the family member ras-related protein 3a (Rab3a), highly abundant in brain tissues, which has been connected with several neurodegenerative diseases (AD, PD, and DLB) due to its regulation of Aβ production and interaction with α-syn. 262 The protein has been investigated by Bereczki et al, 153 which however did not find any significant difference in CSF between patients with PD and control. A second important protein family for neurotransmitter exocytosis is the granin family, which is constituted of dense-core vesicle proteins involved, inter alia, in neuropeptide biogenesis and secretion. The proteins have not only been associated with neurodegenerative diseases, such as AD, but also with other synaptopathies, such as schizophrenia and depression. 263 Three of the key granins: chromogranin-A, secretogranin-2, and neurosecretory protein VGF, have been found to have significantly lower CSF concentrations in AD.231,264
Another synaptic protein involved in the pathology of AD is contactin-2, a cell-adhesion protein that interacts with APP and beta-secretase 1 (BACE1). Chatterjee et al 265 found that the protein was reduced in both brain tissue and CSF in AD. The less well-studied members of the synuclein family, beta-synuclein (β-syn) and gamma-synuclein (γ-syn), are also present in proteinaceous aggregates in some α-synucleinopathies. 266 Oeckl et al 167 was the first to measure all 3 synucleins protein family members, α, β, γ in CSF. They found increased concentrations of all synucleins in AD and CJD; however for PD, DLB, and atypical parkinsonian syndromes the concentrations were not altered. Furthermore, a high correlation between the 3 synucleins was seen. In another study by Oeckl et al, 267 β-syn was quantified in blood and found it to be increased in AD and CJD compared with controls but not in other neurodegenerative diseases, such as PDD, DLB, amyotrophic lateral sclerosis (ALS), and FTD.
14-3-3
14-3-3 proteins refer to a family of 7 isoforms which are highly expressed in the brain, accounting for 1% of its soluble protein content. They are also particularly enriched at synapses (presynaptic) and important modulators of synaptic functions, such as neurotransmission and plasticity. 14-3-3 protein detection by Western blot has since long been used to detect CJD, albeit this technique is only semi-quantitative. However, more recently, 14-3-3 have been studied in the context of other neurodegenerative pathologies. 14-3-3 isoforms have not only been found to co-localize in LB in PD and NFTs in AD, but also been found to interact with key proteins such as tau and α-syn. They have also been genetically linked to both neurodegenerative diseases (PD, AD, and CJD) and neuropsychiatric disorders (schizophrenia and bipolar disorder).268,269 A recent study by Antonell et al 270 found significantly increased gamma 14-3-3 concentrations in both FTD and AD compared with controls. For AD, increased concentrations were found already in a prodromal stage and the protein level was also significantly higher at later stages compared with FTD. Furthermore, when analyzing for 14-3-3, 96% of subjects were positive for neurodegeneration when applying the AT(N) system, compared with 94% for neurofilament light and 62% for neurogranin. 270
Synaptophysin is one of the most used synaptic biomarkers in immunohistochemistry since it is the most abundant integral synaptic vesicle and plasma membrane protein. In studies of AD postmortem brain tissue, it has been shown that the synaptophysin content is reduced.271,272 Several studies have reported that the protein is not detectable in CSF,139,138,273 possibly due to its high hydrophobic profile. 139 However, it has recently been reported to be detected in exosome preparations from body fluids.274,275
Neuronal-derived exosomes
A recent approach for the discovery of new synaptic biomarkers has been based on isolating neuronal exosomes from blood (plasma). As discussed previously, blood is an easily accessible peripheral fluid, preferred to CSF, which entails a more invasive extraction procedure. However, blood has the disadvantage of being further away from the brain and give peripheral contribution to the levels of the protein. Studying neuronal exosomes enriched from blood gives the advantage to use blood while hopefully better reflecting brain pathogenic processes. Explorative proteomic analysis has tried to map the protein content of the neuronal exosomes and confirmed the presence of several synaptic proteins such as Rab3a and GRIA4. 276 In plasma samples, Goetzl et al 274 reported significantly decreased neuronal-derived levels of synaptophysin together with synaptopodin, synaptotagmin-2, and neurogranin in patients with AD and FTD compared with controls. In the same study, GAP-43 and synapsin-1 were also detected, but were found to have significantly lower levels only in AD. Furthermore, in another study by Goetzl et al, 277 plasma neuronal–derived exosome levels of NPTX2, neurexin 2, GRIA4, and neuroligin 1 were found to be significantly decreased in AD, where also GluR4 and neuroligin 1 correlated with cognitive loss. Another protein that has been quantified in neuronal exosomes is α-syn, found to have increased concentrations in PD compared with controls. 278 For proteins such as neurogranin or α-syn, where peripheral expression complicates the quantification in blood, neuronally derived exosomes seem like an excellent option. However, even if this has promise, it is limited by expensive and time-consuming sample preparation, which as of today restricts its potential for high-throughput biomarker screening and its use in clinical routine. Nevertheless, exosomes are being connected to an increasing number of synaptopathies and they have even been implicated in the propagation of disease-associated proteins such as tau, Aβ, PrPC, and α-syn.279,280 They are a relatively unexplored source for synaptic biomarkers, which makes them a vital part of the field (Figure 2).

Proteomic approaches in synaptic biomarkers discovery and validation. Proteomic studies can start with large explorative investigations in brain tissue, which might lead to the discovery of new candidate biomarkers. However, these studies can be seen as starting points, and they have no clinical utilities. Thus, investigations in CSF are needed to be able to translate the biomarker discovery into a tool of clinical use. Once the biomarker has been validated in CSF, further investigations can be carried in blood, a biofluid with higher accessibility and cheaper to use. On the other hand blood is further away from the brain and the targeted protein level might be susceptible to peripheral contribution, resulting in lower biomarker specificity and confounding results. A possible approach to overcome this problem is the use of plasma-derived neuronal exosomes. These investigations can be carried out with a targeted or non-targeted approach. In the diagram, pros and cons of both approaches are highlighted. ELISA indicates enzyme-linked immunosorbent assay; IP, immunoprecipitation; LC-MS, liquid chromatography-mass spectrometry; PRM, parallel reaction monitoring; SIMOA, single molecule array; WB, Western blot.
Conclusions and Future Perspective
Synapses are essential interconnecting points for neurons and are primarily affected in neurodegenerative and neurodevelopmental disorders. 72 Accumulation of misfolded proteins seems to directly affect them, 35 leading to their dysfunction and loss, which is closely related to the cognitive deficits seen in these aging disorders. This review summarizes latest studies on more established and newly investigated synaptic proteins as candidate biomarkers for synapse dysfunction and neuronal injury in different neurodegenerative diseases, in relation to both CSF and blood (Table 1).
Synaptic biomarkers changes in CSF and blood based on current literature.
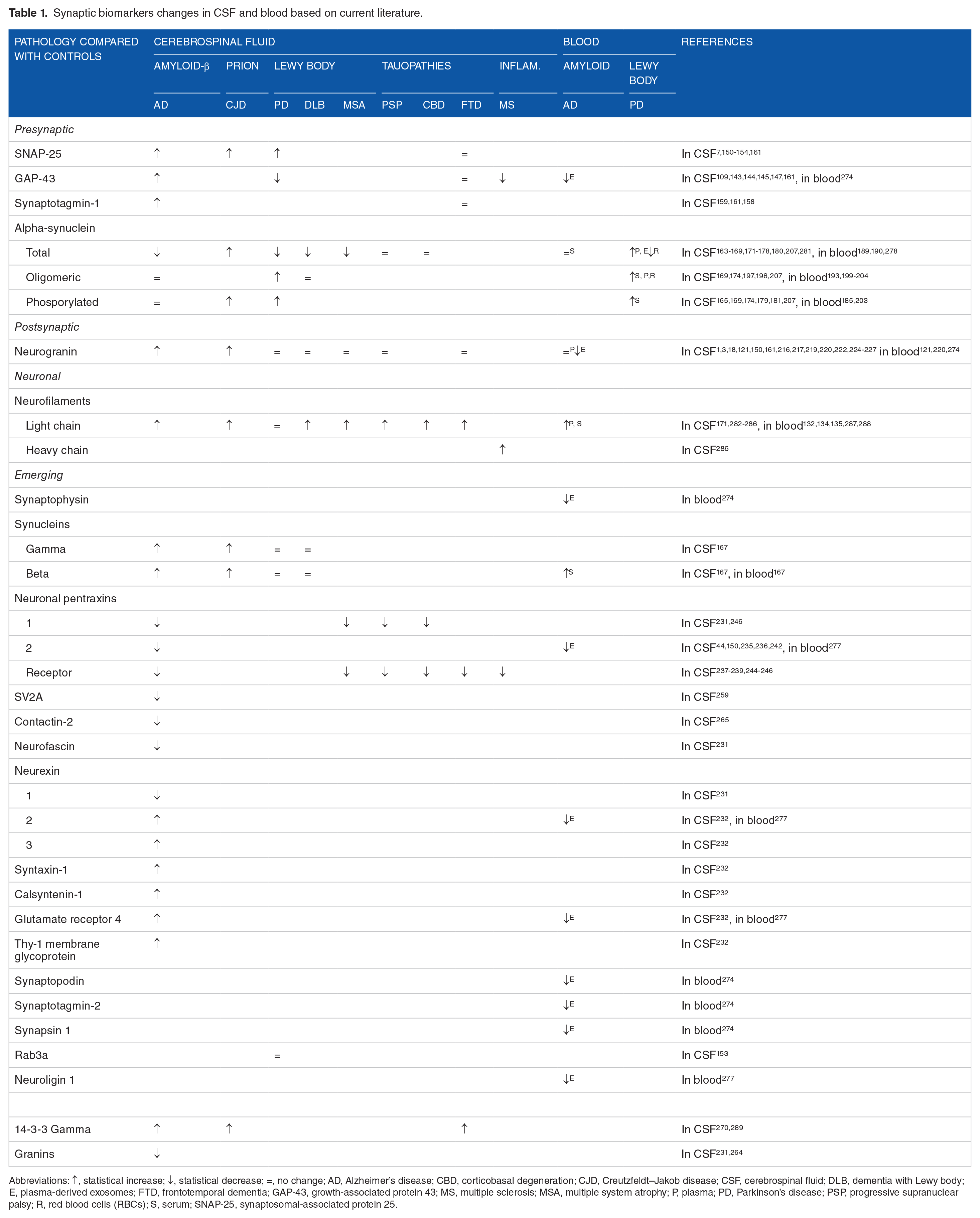
Abbreviations: ↑, statistical increase; ↓, statistical decrease; =, no change; AD, Alzheimer’s disease; CBD, corticobasal degeneration; CJD, Creutzfeldt–Jakob disease; CSF, cerebrospinal fluid; DLB, dementia with Lewy body; E, plasma-derived exosomes; FTD, frontotemporal dementia; GAP-43, growth-associated protein 43; MS, multiple sclerosis; MSA, multiple system atrophy; P, plasma; PD, Parkinson’s disease; PSP, progressive supranuclear palsy; R, red blood cells (RBCs); S, serum; SNAP-25, synaptosomal-associated protein 25.
Current CSF synaptic biomarkers are altered in AD but seemingly not in other neurodegenerative disorders. This can reflect a higher response of synapses and neurons to Aβ-mediated damage, probably making AD the pathology with the highest synaptic damage. However, more efforts are needed to characterize synaptic loss in non-AD dementias and other synaptopathies. Increasing evidence suggests that synaptic dysfunction is also involved in neurodevelopmental diseases290,291 and neuropsychiatric disorders.26,292 Thus, the study of these conditions may help understanding differences or commonalities between synaptopathies. 293
It can be noticed that most of the synaptic biomarkers described are represented by presynaptic proteins 294 and, in AD, glutamatergic synapses appear to be primarily affected.6,294-297 Among the reviewed synaptic proteins, neurogranin is the most extensively studied and the evidence presented thus far is seemingly specific for AD or Aβ deposition. The other synaptic proteins also show changed levels in relation to AD, with most of them showing increased CSF concentrations, but also in non-AD neurodegenerative diseases (eg, PD, tauopathies), even though in these diseases they are less investigated. NfL is a good marker for general neuronal loss and it would be suitable to represent the “N” in the ATN criteria119,298 given that CSF t-tau also mainly changes in AD and CJD. Blood NfL strongly reflects CSF NfL. 299 Elucidating the mechanisms of release of these proteins into biofluids would be of importance to understand their changes in concentration, thus connecting pathological mechanisms to biological responses and increase the interpretability of this biomarker category.
Understanding the pathological mechanisms responsible for synaptic damage is of central importance also during synaptic biomarker investigation. Brain studies could be a starting point, helping to understand the pathophysiological events and for selecting biomarker candidates. The next steps may involve the investigation of biofluids, like CSF, ideally followed by studies in blood, representing the way to bring the investigation further and possibly find synaptic biomarkers of clinical utility. The future of biomarkers ideally would be able to rely on sampling blood, which is a more accessible source than CSF. However, the possible contribution of peripheral expression of the biomarker protein, as discussed for neurogranin and α-syn, can represent a problem and, to date, we still have no blood biomarkers reflecting synaptic pathology. Neuronal-derived exosomes in blood can represent an alternative; however the complexity and variability of the exosome enrichment procedure is currently a drawback for large studies and routine use.
Future directions of research should consider more longitudinal studies, to compare protein time-related changes with the disease progression. The contribution of sex differences should be also considered in more detail, as developing evidence suggests that differing biomarker profiles do exist but is protein-specific.300,301
In conclusion, the available evidence on CSF synaptic biomarkers points toward the possible use of these proteins as indicators of synaptic alteration and elimination in synaptopathies, and their use to follow cognitive deficits in neurodegenerative diseases. More efforts are needed to assess their possible use in blood. Mechanistic studies will possibly help understanding how those proteins are affected in pathological processes thus increasing their value as potential biomarkers. Moreover, developing assays for their quantification using highly sensitive and high-throughput platforms will push synaptic protein quantification toward broader investigations. This overview of the field will hopefully highlight possible gaps and guide future studies.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), and European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236). HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), and the UK Dementia Research Institute at UCL.
Declaration of conflicting interests:
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. HZ has served at scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, and CogRx; has given lectures in symposia sponsored by Fujirebio, Alzecure, and Biogen; and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program.
Author Contributions
EC, JN and NJA provided the initial idea and outline of content of the manuscript; EC created figures 1 and 2. JN created the table. All authors contributed to the content of the article and critically reviewed and edited the manuscript.