
Abstract
Objective:
This study aimed to identify biomarkers for colorectal cancer (CRC) with representative gene functions and high classification accuracy in tissue and blood samples.
Methods:
We integrated CRC DNA methylation profiles from The Cancer Genome Atlas and comorbidity patterns of CRC to select biomarker candidates. We clustered these candidates near the promoter regions into multiple functional groups based on their functional annotations. To validate the selected biomarkers, we applied 3 machine learning techniques to construct models and compare their prediction performances.
Results:
The 10 screened genes showed significant methylation differences in both tissue and blood samples. Our test results showed that 3-gene combinations achieved outstanding classification performance. Selecting 3 representative biomarkers from different genetic functional clusters, the combination of ADHFE1, ADAMTS5, and MIR129-2 exhibited the best performance across the 3 prediction models, achieving a Matthews correlation coefficient > .85 and an F1-score of .9.
Conclusions:
Using integrated DNA methylation analysis, we identified 3 CRC-related biomarkers with remarkable classification performance. These biomarkers can be used to design a practical clinical toolkit for CRC diagnosis assistance and may also serve as candidate biomarkers for further clinical experiments through liquid biopsies.
Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide. There are approximately 1 850 000 new cases and 880 000 deaths from CRC every year. 1 The 5-year survival rate of patients with CRC varies depending on the circumstances; the average 5-year survival rate of patients with in situ carcinoma is greater than 90%, whereas it is less than 20% for patients with metastasis. 2 The main diagnostic methods for CRC include colonoscopy, sigmoidoscopy, computed tomography (CT) colonography, and fecal immunochemical tests (FITs); of these, colonoscopy is the most common. Although colonoscopy has high sensitivity, it has several limitations. First, it may cause discomfort due to its invasive nature. Second, it may cause complications such as gastrointestinal perforation and bleeding. Third, it is expensive and requires bowel preparation before surgery. 3 Sigmoidoscopy can only reveal a part of the gastrointestinal tract and may ignore some abnormal areas. CT colonography carries the risk of radiation exposure and requires bowel preparation. FIT reflects a higher number of false-positive cases than other tests and has relatively low specificity because cancer does not necessarily require FITs. 4
Recent studies have shown that in addition to gene variants, changes in epigenetics, such as DNA methylation, histone modification, chromatin conformation, and microRNAs, are also important factors leading to cancers. 5 Among these changes, DNA methylation has greater stability and reliability across various cellular conditions. 6 DNA methylation is a process that regulates gene expression by adding a methyl group to the fifth position of cytosine through DNA methyltransferases (DNMTs) to form 5-methylcytosine without changing the DNA sequence. 7 Compared to the irreversible characteristic of genetic mutations, DNA methylation is a reversible process that can be inhibited by certain drugs to inactivate DNMTs. 8 Therefore, interventions that may alter DNA methylation are potential therapies for various cancers.
Aberrant DNA methylation is associated with several diseases, including cancer, autoimmune diseases, metabolic syndromes, and neurological disorders. 9 There are 2 patterns of DNA methylation: hypermethylation and hypomethylation. Hypermethylation typically occurs at CpG sites near promoters, suppressing gene expression. Silencing tumor suppressor genes through hypermethylation can increase the risk of cancer development. In contrast, hypomethylation typically occurs outside CpG islands, leading to the upregulation of gene expression. 10 DNA methylation microarrays have several advantages compared to other analytical methods, including lower cost and amount of DNA required. Traditionally, tissue samples have been used to analyze DNA methylation during cancer detection; however, this technique has several limitations. First, acquiring tissue specimens requires invasive sampling, and the location of the tumor can affect the difficulty in acquiring samples, thereby increasing the risk of sampling. In addition, issues regarding the quality of surgically resected tissues may affect the experimental results. 11
Circulating tumor DNA (ctDNA) is a type of cell-free DNA (cfDNA) that consists of DNA fragments derived from tumor cells in the blood. When tumor cells rupture or undergo apoptosis, DNA fragments are released into body fluids, primarily the blood. 12 Compared to tissue biopsies, cfDNA methylation testing requires only a blood sample from a subject, without the need for any invasive surgical procedures. Moreover, cfDNA methylation testing has potential advantages for early cancer detection because it can detect the methylation status of specific genes even before tumorigenesis occurs.
Most previous studies have been limited to using only one type of sample, such as tissue, blood, or stool, to identify biomarkers associated with CRC. In this study, we aimed to identify biomarkers with high accuracy in both tissue and blood samples. Specifically, genome-wide methylation and comorbidity association analyses were performed to identify candidate biomarkers associated with CRC. Biomarkers suitable for methylation in clinical trials were selected to verify the differential gene methylation status between patients with cancer and normal controls. Finally, several machine learning methods were used to select specific biomarkers from various functional gene clusters for testing. We used tissue samples as the training dataset and included blood samples in the testing dataset to evaluate the classification accuracy of the selected biomarkers.
Materials and Methods
Dataset description
In this study, CRC DNA methylation profiles from The Cancer Genome Atlas (TCGA) were used for genome-wide methylation analyses and machine learning model training. The dataset comprised 314 patients with CRC and 38 normal subjects. Additionally, cfDNA methylation profiles from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo) database were used to validate the selected biomarkers. The GSE122126 dataset consisted of cfDNA blood samples from 4 subjects with CRC, 3 with breast cancer subjects, 4 with lung cancer, and 30 normal subjects. 13 The GSE129374 dataset consisted of cfDNA blood samples from 22 pairs of patients with liver cancer and normal controls. 14 The GSE220160 and GSE274189 datasets consisted of 8 and 12 patients with CRC, respectively. All data from the TCGA database and the GSE122126 and GSE129374 datasets from the GEO database were derived from the Illumina Infinium HumanMethylation450 (450 K) BeadChip array, covering 485 577 CpG sites. Data from the GSE220160 and GSE274189 datasets were derived from the Illumina Infinium HumanMethylationEPIC BeadChips, covering 868 565 CpG methylation sites for each array. Both BeadChips platforms are known for storing and accessing methylation profiles, which are saved as IDAT files.
For experimental validation, 30 paired normal and CRC tissue samples were preserved in 10% neutral-buffered formalin after surgical excision (IRB: C202105205). Each sample consisted of 2 tissue sections less than 3 × 2 cm in size. All tissue samples were processed to create formalin-fixed paraffin-embedded (FFPE) specimens, which were subsequently subjected to DNA extraction. The total amount of DNA extracted from the normal tissues ranged from 120 to 5548 ng, whereas the total amount of CRC tumor-isolated DNA ranged from 192 to 13 480 ng.
Candidate biomarker selection
This study utilized the R package ChAMP to import methylation profiles in the IDAT format. 15 First, the data preprocessing procedures included filtering out the methylation probes unsuitable for analysis, and Beta-Mixture Quantile Normalization (BMIQ) was subsequently performed. 16 Due to experimental noise, the distribution of beta values for certain CpG sites was not concentrated. To address this issue, we applied the interquartile range (IQR) method to identify and eliminate outliers in beta-values at each CpG site. Any values that were more than 1.5 IQR below Q1 or more than 1.5 IQR above Q3 were considered outliers. Next, the beta value difference (Δβ) between the experimental group (subjects with cancer) and the control group (normal subjects) was calculated. Adjusted p-values and Δβ thresholds were also set to identify differentially methylated positions.
Some potential diseases associated with tumor formation and progression may occur before carcinogenesis. 17 Therefore, the comorbidities highly correlated with CRC were determined and analyzed to enhance the detection potential of candidate biomarkers. The biomarkers associated with CRC and its comorbidities were selected from DisGeNet, the largest comprehensive database containing information on human disease-associated genes and variants. 18
Gene functional clustering analysis
We classified candidate biomarkers near promoter regions into multiple functional clusters based on the functional similarity between genes to select representative and functionally diverse biomarkers. Gene Ontology (GO) representation was used to systematically annotate the functions of each gene. Each GO term represented a specific function and was classified into one of the following 3 categories: cellular components (CC), molecular functions (MF), or biological processes (BP). These terms are organized into a directed acyclic tree structure.
Although not all genes were comprehensively annotated, most of them and their corresponding functions were assigned at least 1 GO annotation. Before the similarity between 2 genes was calculated, the similarity of all pairs of GO terms was calculated. The algorithm used to calculate GO term similarity was based on semantic similarity, 19 and can be divided into 2 categories: information content-based methods and graph-based methods. 20 The Python package GOntoSim utilized in this study is based on the graph-based approach. 21 In contrast to the other methods, GOntoSim considers both the lowest common ancestor and the common descendants of 2 GO terms to calculate both upward and downward similarities. 21 This approach can fix errors arising from the node depth.
The similarity between 2 genes was obtained by calculating the similarity across all GO term pairs. For example, if there were 2 genes, A and B, their GO terms were divided into 3 categories: CC, BP, and MF. The GO similarity for each category was calculated using equation (1), where GOAi and GOBj are the ith GO term of gene A and the jth GO term of gene B, respectively, and m and n are the numbers of GO terms of genes A and B, respectively. The overall similarity between the genes A and B was calculated using equation (2), where #BP, #CC, and #MF are the numbers of GO terms belonging to BP, CC, and MF, respectively, and SimBP, SimCC, and SimMF are the GO similarities of BP, CC, and MF, respectively.

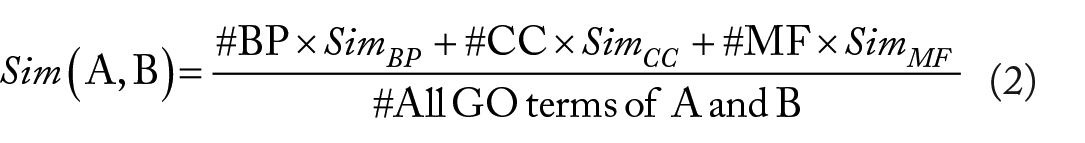
To facilitate functional clustering, we represent the similarities between all genes as a distance matrix. The greater the similarity, the closer the distance between 2 genes, representing a greater correlation between their functions. Ward’s method was used to cluster genes based on their distances, which reduced the variability within each gene cluster.
Clinical validation of the selected DNA methylation biomarkers
The DNA of 30 paired normal and CRC tumor samples were extracted from FFPE tissue specimens. These samples were subjected to bisulfite conversion using an EZ DNA Methylation-Lightning™ kit (Zymo Research, Cat. #D5031) following the manufacturer’s instructions. Next, the bisulfite-converted DNA was subjected to quantitative polymerase chain reaction (qPCR), which was performed on an Applied Biosystems QuantStudio™ 6 Flex Real-Time PCR System using the standard SYBR green method and methylation-specific primers to determine the methylation levels via pre-defined calibration curves. The primer sequences used in the quantitative methylation-specific PCR (qMSPCR) assay are listed in Table 1.
The sequences of primers used in the qMSPCR assay.

qMSPCR is typically used to detect hypermethylated promoter genes (HPGs). 22 Hypomethylated loci possess variable features with relatively low levels of methylation. Hence, we selected only hypermethylated genes as candidate biomarkers to design primers for clinical qRT-PCR experiments.
Furthermore, we compared the methylation levels of the loci used in the clinical experiments with our in silico methylation analytical results to ensure consistent degrees of methylation. Both beta and M-values were used to evaluate the methylation levels of the selected loci, as these 2 values provide different perspectives on the methylation status of genes. Studies have shown that the M value method provides better statistical power, whereas the beta value has more biological significance.23,24
Classification testing of gene combinations
To identify methylation biomarkers that can accurately detect CRC and gene combinations that maintained consistency in methylation status between tissue and blood samples, 3 machine learning methods, namely, support vector machine (SVM), random forest, and logistic regression, were applied. Tissue samples from TCGA were utilized as training datasets and 5-fold cross-validation was performed. Next, the samples from the GEO database were used as the testing datasets to validate the classification accuracy. Since we expected that our selected biomarkers would demonstrate specificity for CRC, the datasets from other cancers (breast, lung, and liver cancer) were included to ensure that our model can distinguish CRC from these other cancers. During the testing procedure, we regarded the CRC subjects as the experimental group (labeled as 1) and the subjects with other cancers, along with the normal subjects, as the control group (labeled as 0).
According to the practical probe design for qMSPCR experiments, the number of genes within the testing toolkit should be limited. We aim to use as few genes as possible to achieve a high detection performance. In addition, we considered selecting a representative gene from a specific functional group to avoid selecting redundant genes with similar biological functions or within the same biological pathway. The selected HPGs were classified into different functional groups and gene combinations containing different numbers of genes were generated. Each combination was evaluated using different machine learning models. We used the F1 score and MCC to evaluate the classification performance of each combination. The combination with the highest average classification metrics was subsequently selected, and the representative biomarkers from the proposed procedures were considered the target biomarkers for CRC diagnosis.
Results
Significantly different DNA methylation biomarkers and comorbidity-related genes for CRC
To screen for genes with significant differential methylation and associated CRC comorbidities, we filtered 485 512 CpG sites using a quality control procedure, 15 and 241 088 CpG sites remained after filtering. Next, Δβ and false discovery rate (FDR)-adjusted p-value thresholds (FDR-adjusted P-value < .01; |Δβ| ⩾ .5) were set, and 599 CpG sites met the threshold criteria (525 hypermethylated CpG sites and 74 hypomethylated CpG sites); these loci were subsequently mapped to 252 genes. These genes were defined as significantly differentially methylated candidate biomarkers.
The significant comorbidities of CRC, including hemorrhage of the gastrointestinal tract, hemorrhoids, constipation, duodenal ulcers, peptic ulcers, unspecified functional disorders of the stomach, and abdominal pain, were identified from the literature.25-30 We then identified the genes associated with CRC and its comorbidities using DisGeNet. The number of genes associated with each comorbidity is shown in Table 2.
Comorbidities of colorectal cancer.

After intersecting significantly differentially methylated biomarkers and CRC comorbidity-related genes, 141 genes remained, of which 42 hypermethylated genes were located near promoter regions. As tumor suppressor genes located near promoter regions can easily cause genetic abnormalities and, in turn, lead to carcinogenesis, 31 we performed further functional analyses of these candidate genes.
Functional clustering analysis of HPGs
A similarity matrix of the 42 HPGs was obtained by calculating pairwise gene-gene distances (Figure 1). We utilized Ward’s hierarchical clustering method to classify the HPGs, and the clustering results are graphically presented as a dendrogram in Figure 2. For the practical design of the testing kit, the 42 HPGs were divided into 2 to 4 clusters. In addition, we queried the DAVID database 32 to identify the predominant GO annotation features within each functional cluster. The first cluster was characterized by the following GO terms: BP, regulation of transcription from the RNA polymerase II promoter; CC, nucleus; MF, RNA polymerase II transcription factor activity, and sequence-specific DNA binding. The second cluster included GO terms such as adult locomotor behavior (BP), plasma membrane (CC), and receptor binding (MF). The third cluster included GO terms such as extracellular matrix organization (BP), extracellular matrix (CC), and metallopeptidase activity (MF).

Similarity matrix of the 42 hypermethylated promoter genes identified in this study.

Dendrogram of the hierarchical clustering result of the 42 HPGs identified in this study.
Clinical validation of the target biomarkers
Only 20 candidate loci were chosen from the 42 initial HPGs for methylation-specific primer synthesis based on 3 criteria: (1) high accuracy and adjusted P-value in distinguishing tumor specimens from normal samples, (2) candidates from each functional cluster were included based on the proportions of the gene counts, and (3) any predicted non-specific primers or primers with dimers or hairpin issues were excluded.
In addition, primer performance tests were conducted with 20 methylation-specific primer pairs using 0%, 50%, and 100% methylated DNA. Our results demonstrated that only 10 primer pairs successfully amplified specific PCR products under 50% and 100% methylated DNA conditions. Thus, only 10 candidate CRC biomarkers were screened for further clinical validation.
After performing qMSPCR on 30 patients with CRC, the CT methylation values were used to estimate the degree of methylation in each patient. In addition, differences in methylation between the experimental group (patients with CRC) and the control group (normal subjects) were determined. Lower methylation CT values indicate a lower number of PCR cycles required to reach a specific methylation threshold, indicating a greater degree of methylation. In other words, methylation CT values showed an inverse correlation with the degree of methylation. Figure 3A shows that subjects in the control group generally had higher methylation CT values for the 10 target biomarkers. If outliers were excluded, all genes except IRF4 showed significant differences in methylation. To enhance the reliability of the clinical experiments, we extracted the beta- and M-values of TCGA CRC subjects for the 10 target biomarkers (Figure 3B and C). We found that all biomarkers exhibited significant methylation differences (1 way ANOVA test P < .01).

(A) Boxplot of the CT values for the 10 target biomarkers in patients with CRC for clinical validation. (B) Boxplot of the beta values obtained for the 10 target biomarkers in subjects with CRC in TCGA. (C) Boxplot of the M-values obtained for the 10 target biomarkers in subjects with CRC in TCGA.
Optimal gene combinations
To screen for functionally representative biomarkers with good predictive performance, we selected 1 gene from each functional cluster to form various gene combinations. Hence, the total number of combinations depended on the defined number of functional clusters. There were 9 gene combinations if only 2 functional groups were defined, 20 if there were 3 defined functional groups, and 24 if there were 4 defined functional groups. To determine the best combinatorial number of candidate genes, we compared the classification performance of the 2-, 3-, and 4-gene combinations using the SVM, random forest, and logistic regression prediction models. The results of the SVM or logistic regression prediction models showed that the average F1 score and MCC of all 3-gene and 4-gene combinations were approximately 0.9. For the random forest model, there was only one 2-gene combination with an F1 score of over 0.9; five 3-gene combinations and seven 4-gene combinations had F1 scores of over 0.9. These results showed that adding a fourth gene did not significantly improve the overall test performance. Therefore, the 3-gene combination was considered the most effective predictor of CRC diagnosis in this study.
Boxplots were then used to represent the test performance (MCC and F1 score) for the twenty 3-gene combinations obtained (Figure 4). The gene combination with the best performance comprised ADHFE1, ADAMTS5, and MIR129-2. The best MCC and F1 scores of the 3 machine learning methods were 0.92 and 0.94 for SVM, 0.87 and 0.90 for random forest, and 0.92 and 0.93 for the logistic regression model, respectively.

Boxplot of the test results for 20 gene combinations. The x-axis represents the F1 score and MCC of the 3 machine learning methods, while the y-axis indicates the metric values. Each point in the plot represents a gene combination.
Discussion
DNA methylation has recently become the most promising and effective method for cancer diagnosis. For asymptomatic patients at different stages of cancer, biomarkers with significantly different methylation and associations with specific comorbidities can be used for cancer diagnosis, which enables them to receive proper prognostic treatment and thereby increasing their survival rate. In this study, we performed a differential methylation analysis of CRC samples and initially screened 252 genes with notable methylation differences. Among these, 141 were associated with various CRC comorbidities. After gene selection and applicability screening for hypermethylation characteristics, 10 genes (ADHFE1, ADARB2, EFS, ADAMTS5, MMP23B, PLD5, MIR129-2, IRF4, NRG1, and KCNQ5) were selected for clinical validation using qMSPCR. The results showed that these 10 genes had significant differences in CT values in CRC tissue samples. To meet the requirements of the diagnostic kits and limit the number of methylation biomarkers, we performed gene functional analysis to select representative genes from each functional group. Next, functionally representative biomarker combinations were selected to construct prediction models. Twenty biomarker combinations were assembled into 3 functional clusters. Our results showed that the 3-gene combination ADHFE1, ADAMTS5, and MIR129-2 provided the best predictive performance among the 3 machine learning models employed, with MCCs greater than 0.85 and F1 scores greater than 0.9.
Recently, the use of epigenome-wide DNA methylation data to identify methylation biomarkers associated with specific cancers has gained popularity. For instance, Shen et al identified 2 CRC biomarkers (SDC2 and SHOX2) associated with precancerous lesions using qMSPCR and validated them using blood and stool samples. 33 Chang et al identified the TMEM240 gene and showed that this hypermethylated gene can repress cancer cell proliferation and migration. 34 In the study by Baharudin et al, 35 10 HPGs were identified through DNA methylation analysis of 54 paired tumor–adjacent normal colon samples from Malaysian subjects using the 450k assay. Among these, 5 HPGs including ADHFE1, USP44, ADARB2, ZNF582, and ZNF132 also presented in our list of 42 HPGs. However, our functional clustering analysis revealed that these 5 HPGs were clustered in the same functional group, suggesting potential genetic function redundancy. Furthermore, half of the 10 HPGs belong to the ZNF gene family, and 4 of them also appeared in our 42 HPGs. Previous studies have shown that ZNF family genes are significantly associated with CRC development, acting as either oncogenes or tumor suppressor genes.36,37 In the study by Muthamilselvan et al, 38 27k CRC methylation data from TCGA were profiled, and 7 hypermethylated stage-salient genes in promoter regions were identified. Our results showed that while these genes exhibited significantly differential methylation, they were not associated with CRC comorbidities. Therefore, these genes were not included in our final biomarker list. These findings suggest that incorporating genes related to CRC comorbidities could provide a substantial impact on the identification of candidate biomarkers.
Among the selected methylation biomarkers, ADHFE1 is a common CRC biomarker. Our results showed that biomarker combinations containing ADHFE1 had excellent performance, indicating that ADHFE1 is important for enhancing the overall performance of biomarker combinations. Moon et al and Hu et al reported that the hypermethylation of ADHFE1 promotes the proliferation of CRC cells.39,40 Furthermore, Tae et al reported that the hypermethylation of ADHFE1 is associated with CRC cell differentiation. 41 In addition, we identified several biomarkers known to be associated with CRC (ADAMTS5 and KCNQ5). Li et al reported that ADAMTS5 can inhibit the migration and invasion of CRC cells but has no effect on cell growth or apoptosis. 42 Gao et al indicated that the methylation of the MIR129-2 promoter CpG islands may lead to dysregulation of methylation in CRC. 43
Compared with the present studies which perform in silico DNA methylation analysis to select CRC biomarkers, our study includes multiple test datasets containing normal subjects and subjects with other cancer types. In addition, we performed clinical experiments to ensure that the methylation status of our target biomarkers is consistent across both in silico DNA methylation analysis and clinical experiments. Although our study included only a small amount of cfDNA samples in the testing dataset, the results showed that the 10 biomarkers exhibitdc significant methylation differences in these samples, indicating that these biomarkers may play as potential candidates for clinical experiments on cfDNA samples from CRC subjects. Our test results suggest that the selected biomarkers either have high robustness and specificity for CRC or demonstrate excellent classification performance.
Conclusion
In this study, we performed an integrated DNA methylation analysis and identified effective CRC biomarker combinations that showed significantly different hypermethylation in both tissue and blood samples, suggesting their potential as candidate biomarkers for further clinical experiment for CRC subjects using liquid biopsies. These biomarkers were strongly associated with certain CRC comorbidity patterns and demonstrated high accuracy in CRC detection. Notably, our results showed that the biomarker combination ADHFE1, ADAMTS5, and MIR129-2 achieved the best classification performance across the 3 machine learning models employed. Because of budget constraints, we only included 30 clinical samples for preliminary experiments. Nonetheless, we expect that the selected biomarkers will be used to detect CRC in a larger cohort in the near future.
Footnotes
Acknowledgements
Abbreviations
CRC Colorectal Cancer
CT Computed Tomography
FIT Fecal Immunochemical Test
DNMT DNA methyltransferase
ctDNA Circulating tumor DNA
cfDNA Cell-free DNA
TCGA The Cancer Genome Atlas
GEO Gene Expression Omnibus
FFPE Formalin-fixed paraffin-embedded
ChAMP Chip Analysis Methylation Pipeline
BMIQ Beta-Mixture Quantile Normalization
DMPs Differentially Methylated Positions
GO Gene Ontology
CC Cellular Component
MF Molecular function
BP Biological Process
LCA Lowest Common Ancestor
FFPE formalin-fixed, paraffin-embedded
qPCR quantitative polymerase chain reaction
qMSPCR quantitative Methylation-specific Polymerase Chain Reaction
HPGs Hypermethylated promoter genes
SVM Support Vector Machine
MCC Matthews correlation coefficient
DAVID Database for Annotation, Visualization and Integrated Discovery
PCR Polymerase Chain Reaction
Author Contributions
Conceptualization, Y.-H.T. and T.-W.P.; methodology, Y.-H.T., Y.-C. C., and T.-W.P.; software, Y.-H.T.; validation, Y.-H.T. and Y.-H.L.; formal analysis, Y.-H.T. and Y.-H.L.; investigation, S.-J.C., Y.-C. C., and T.-W. Pai; resources, S.-J.C. and Y.-C. C.; data curation, Y.-C. C. and T.-W.P.; writing—original draft preparation, Y.-H.T.; writing—review and editing, Y.-H.L., Y.-C. C., and T.-W.P.; visualization, Y.-H.T. and Y.-H.L.; supervision, Y.-C. C. and T.-W.P.; project administration, Y.-C. C. and T.-W.P.; funding acquisition, S.-J.C., Y.-C. C., and T.-W.P. All authors have read and agreed to the published version of the manuscript.
Data Availability
The datasets generated during and/or analyzed during the current study are available in the following repositories: The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/) and Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122126, ![]() ,
,
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Science and Technology Council (NSTC 111-2622-E-027-021 and MOST 111-2221-E-027-113-MY2); and Tri-Service General Hospital (TSGH-E-112281 and MND-MAB-D-114077).
Ethics Approval and Consent to Participate
All subjects provided informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Code of Ethics of the World Medical Association Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Tri-Service General Hospital in Taiwan (IRB No. C202105205).
Consent for Publication
Not applicable.