
Abstract
Objectives:
Under the classification of multicategory survival outcomes of cancer patients, it is crucial to identify biomarkers that affect specific outcome categories. The classification of multicategory survival outcomes from transcriptomic data has been thoroughly investigated in computational biology. Nevertheless, several challenges must be addressed, including the ultra-high-dimensional feature space, feature contamination, and data imbalance, all of which contribute to the instability of the diagnostic model. Furthermore, although most methods achieve accurate predicted performance for binary classification with high-dimensional transcriptomic data, their extension to multi-class classification is not straightforward.
Methods:
We employ the One-versus-One strategy to transform multi-class classification into multiple binary classification, and utilize the overlapping group screening procedure with binary logistic regression to include pathway information for identifying important genes and gene-gene interactions for multicategory survival outcomes.
Results:
A series of simulation studies are conducted to compare the classification accuracy of our proposed approach with some existing machine learning methods. In practical data applications, we utilize the random oversampling procedure to tackle class imbalance issues. We then apply the proposed method to analyze transcriptomic data from various cancers in The Cancer Genome Atlas, such as kidney renal papillary cell carcinoma, lung adenocarcinoma, and head and neck squamous cell carcinoma. Our aim is to establish an accurate microarray-based multicategory cancer diagnosis model. The numerical results illustrate that the new proposal effectively enhances cancer diagnosis compared to approaches that neglect pathway information.
Conclusions:
We showcase the effectiveness of the proposed method in terms of class prediction accuracy through evaluations on simulated synthetic datasets as well as real dataset applications. We also identified the cancer-related gene-gene interaction biomarkers and reported the corresponding network structure. According to the identified major genes and gene-gene interactions, we can predict for each patient the probabilities that he/she belongs to each of the survival outcome classes.
Keywords
Introduction
Precision medicine represents a cutting-edge approach to disease prevention and treatment by considering individual differences in genetics, environment, and lifestyle. In this context, cancer classification using microarray gene expression profiling has garnered significant attention.1,2 Many statistical and machine-learning techniques have been applied to binary cancer classification using gene expression data.3,4 These methods include multiple logistic regression models (MLRs), support vector machines (SVMs), K-nearest neighbors (KNNs), linear discriminant analysis (LDA), and random forests (RFs), among others. While machine learning methods are popular, they often face a major drawback: they can be difficult to interpret and may not provide direct estimates of outcome probabilities. In contrast, statistical MLRs offer both explanatory power and probabilistic estimates.
Over the past decade, multicategory classification problems have become a significant focus for biologists and computer science researchers.5 -7 In precision medicine, multicategory classification of cancer patients’ survival outcomes is particularly crucial.8,9 By employing multicategory classification, it is possible to achieve more accurate diagnoses of cancer survival outcomes, which, in turn, enables the development of more tailored and effective treatment options for patients.
The complexity of cancer development is well acknowledged, frequently involving multiple biomarkers that interact synergistically, such as gene-environment (G-E) or gene-gene (G-G) interactions. 10 Therefore, in addition to primary genetic (G) or environmental (E) factors, interacting biomarkers can significantly influence cancer diagnosis. Incorporating these crucial interacting biomarkers into cancer classification models may improve their predictive accuracy.11,12 However, identifying gene-gene (G-G) interactions is challenging due to the ultrahigh dimensionality of transcriptomic data. One approach to address this challenge is to utilize biological network information to help pinpoint genuine G-G interactions. 13 Additionally, another challenge is that gene expression data are often contaminated by outliers.
In high-dimensional statistical learning, regularized regression methods are commonly recommended. 14 However, a notable drawback of this approach is that the model size might exceed the sample size, potentially leading to suboptimal statistical power. 15 To address this issue, it is widely recognized that preliminary feature screening can significantly improve the effectiveness of model selection using regularization methods. Wang and Chen, 16 along with Wang et al, 17 developed overlapping group screening (OGS) methods aimed at identifying active gene-gene (G-G) and gene-environment (G-E) interactions. These methods incorporate gene pathway information and use the identified features to build a survival time prediction model. The OGS approach has also been applied to clinical cancer versus normal outcome classification using a binary logistic regression model. 4 The OGS methods are especially effective in tackling the challenge of a feature set that greatly exceeds the sample size, particularly when the feature groupings (pathways) overlap.
In this study, inspired by the methodology described by Feng et al, 9 we integrate survival indices with clinical characteristics to classify three distinct survival outcome categories: dead with no tumor, dead with tumor, and alive. This classification is applied to cancer transcriptomic data from The Cancer Genome Atlas (TCGA), specifically for kidney renal papillary cell carcinoma (KIRP), lung adenocarcinoma (LUAD), and head and neck squamous cell carcinoma (HNSCC). For example, the TCGA KIRP transcriptomic dataset includes 275 subjects, with 235 (85.5%) alive, 12 (4.4%) dead with no tumor, and 28 (10.0%) dead with tumor. The dataset is inherently “imbalanced” in nature.
A dataset is termed “imbalanced” when certain classes have significantly fewer subjects compared to others. This imbalance can distort classification accuracy, resulting in poor performance for minority classes despite high accuracy in majority classes. Consequently, classification models trained on imbalanced data are at a higher risk of severe overfitting and bias issues. 18 The problem of class imbalance is more pronounced in multi-class classification than in binary classification.
Several strategies have been proposed to address the class imbalance problem and develop accurate prediction models.19,20 In this study, we focus on resampling methods, which fall into 2 main categories: over-sampling and under-sampling. Over-sampling methods involve creating synthetic samples to increase the number of instances in the minority classes. The advantage of over-sampling is that it preserves all original information, but it can lead to overfitting since it involves duplicating data from minority classes. This issue can be mitigated through techniques like cross-validation. Under-sampling methods, on the other hand, reduce the number of instances in the majority classes to balance the dataset. This approach is more effective when there is a large amount of data and the minority class is not excessively small. However, it risks losing valuable data from the majority classes, which is a significant drawback. In this work, we employ over-sampling methods to address the class imbalance issue in TCGA transcriptomic data applications.
In this study, we apply the Overlapping Group Screening (OGS) method to TCGA cancer data with multiple (>2) survival outcomes. Specifically, the OGS technique is used to identify critical transcriptomic features and gene-gene (G-G) interactions associated with these survival categories. Based on these insights, we construct microarray-based cancer diagnosis models. Unlike the traditional binary logistic regression model used by Wang and Chen, 4 we employ multinomial logistic regression to handle the multicategory outcomes. Additionally, we address the challenges posed by the ultra-high dimensionality of the gene expression data, contamination by outliers, and imbalanced outcome classes. We conduct a series of simulations to compare the performance of several machine learning methods (SVMs, LDA, RFs, and KNNs) and a penalized multinomial logistic regression model with a grouped lasso penalty against our proposed method in accurately distinguishing clinical survival samples. We apply the OGS method to TCGA cancer transcriptomic data to identify significant gene-gene (G-G) interactions associated with clinical survival categories. Based on these identified interactions, we then construct microarray-based cancer diagnosis models.
Methods
Data structure and the multiple pathways
Given a multiple (K + 1)-class data with n subjects, where
Random oversampling example (ROSE) for imbalanced data
A popular over-sampling scheme for dealing with imbalanced data is random oversampling example (ROSE) proposed by Menardi and Torelli. 23 The ROSE procedure is a sampling method based on data synthesis, which addresses the problem of class imbalance by generating artificial data from a few minority classes. It recommends using model estimation and evaluation to create a more balanced data, where model evaluation is performed using a smoothed bootstrap re-sampling to validate the chosen estimation technique. The ROSE procedure can be implemented by the R package “ROSE,” and can be naturally applied to the class imbalance problem in multi-class classification.
Evaluation criteria for multicategory classification
Some multicategory classification evaluation criteria are used. Let the recall
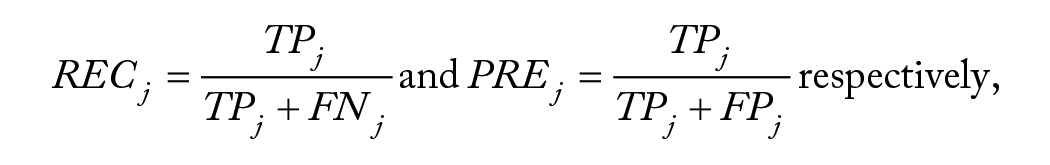
where

Liu et al 24 proposed the overall accuracy (OA) measure, defined as
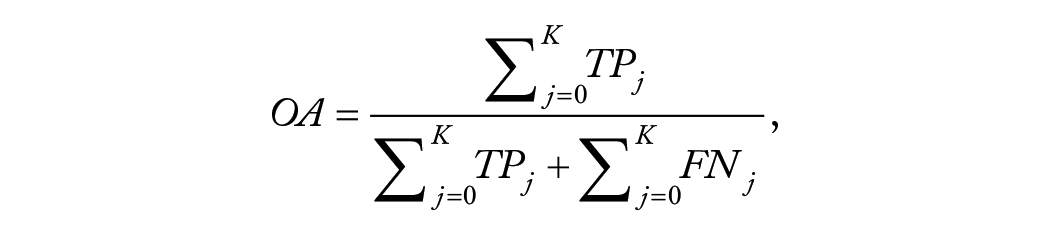
which measures the fraction of correctly classified samples over all samples, and is dominated by the performance in the majority classes. In addition, consider

and

and the macro-F-measure is defined as
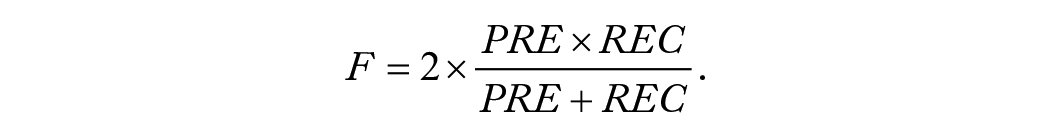
In principle, higher values of REC, PRE, and F reflect better performance of the method, and, in contrast to OA, these metrics reflect more performance in the minority classes.
The overlapping group screening (OGS) approach for binary classification
Here we briefly review the OGS for binary classification in Wang and Chen. 4 This procedure involves a two-stage group screening process aimed at identifying main and interaction effects for binary classification. Considering that gene pathways may overlap with each other, that is, different pathways may share common genes, the latent effect approach proposed by Jacob et al 25 is used to consider overlapping group information. We give a simple example in the appendix to illustrate the latent effects approach, which expresses the characteristic effect of the genes as the sum of group-specific effects. All transcriptomic signatures need to be standardized before OGS methods can be applied. The procedure of the OGS method for binary logistic regression models is as follows.
Step 1: We utilize the overlapping group binary logistic regression model to identify important gene groups (pathways) by executing the R package “grpregOverlap.” 26 At this stage, assume that P candidate pathways are identified among all S pathways.
Step 2: We follow the idea of Wang and Chen 16 to construct groups of G-G interaction pairs within a candidate pathway, between 2 distinct candidate pathways identified in Step 1, and between a pathway identified in Step 1 and an uncharacterized pathway. The Sequence Kernel Association Test (SKAT) by Wu et al 27 is then applied to the binary outcomes to derive group-specific P-values for each group of G-G interactions. The SKAT statistic under the binary logistic regression model is defined as

where

where
Under the null hypothesis, it is assumed that all gene-gene interaction pairs in candidate pathway ℎ have no effect. The SKAT statistic for each G-G interaction pairs group follows a weighted sum of chi-square distribution. The group-specific P-value is obtained from the above chi-square distribution using the Davies and Algorithm 28 method, which can be computed by the R package “CompQuadForm.” 29 A smaller P-value indicates greater significance, thereby granting higher priority in selection.
Step 3: We adopt the approach outlined by Wang et al
17
to randomly permute the original data, creating permuted data that adhere to the null model. Re-run Step 2 to calculate the group-specific P-values
The extension of the OGS approach for binary classification to multicategory classification
Without loss of generality, we take class 0 as the reference and then consider the multicategory logistic model
Binary classifiers are commonly used in machine learning to develop classification rules for multi-class problems. 32 One approach to applying binary classification algorithms to multi-class scenarios involves dividing the multi-class dataset into several binary-class datasets and fitting a binary classification model to each subset. This approach includes 2 main strategies: One-versus-Rest (OvR) and One-versus-One (OvO). In the OvR strategy, all classes except the one under consideration are combined into a single class, while in the OvO strategy, the model is trained to distinguish between 2 classes at a time, with each class being compared against the other classes.
The One-versus-One (OvO) strategy offers the advantage of faster training speed because each classifier is trained on data from only 2 classes, making the training process quicker than when training on all classes simultaneously. Additionally, OvO can achieve higher classification accuracy as each classifier focuses specifically on distinguishing between 2 classes. On the other hand, the advantage of the OvR strategy lies in the reduced number of classifiers required only (K + 1) classifiers need to be trained, with each classifier tasked to distinguish one class from all others. The implementation process is relatively straightforward, as it involves constructing (K + 1) binary classifiers and comparing the output of each classifier. However, its disadvantage is that each classifier may face imbalanced datasets during training, which could affect its performance. Moreover, OvR may not achieve the same high accuracy as OvO, as each classifier needs to distinguish all other classes, which can be challenging.
Following Li et al,
33
we adopt the OvO binary classifier to split a multi-class dataset into multiple binary-class datasets, and fit K individual binary logistic models to model the probability ratio of class j to class 0, j = 1,. . .,K. Accordingly, the OGS approach can be extended naturally to multinomial logistic regression models for multicategory survival outcomes in cancer diagnosis via the OvO strategy of Li et al.
33
Specifically, we divide the whole dataset into K datasets
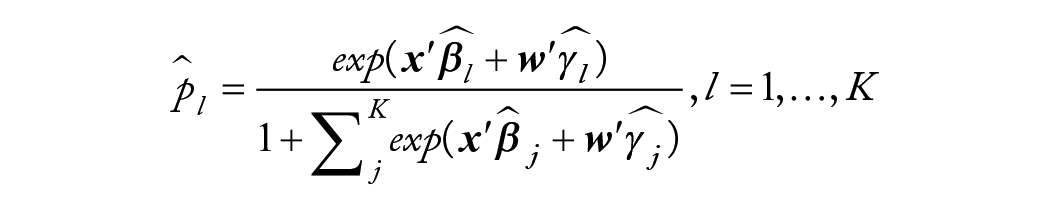
and
The alternative classification methods
The “SIS_GROUP_LASSO” method utilizes a two-stage selection procedure, 34 where the top n/(2∙log(n)) main predictors are selected in the first step by univariate multinomial logistic regressions with the marginal Akaike information criterion (AIC), and in the second step, we examine the interactions corresponding to the main effects selected in the first step. Then, the penalized multinomial logistic regression model with a grouped-lasso penalty for all the K + 1 coefficients (corresponding to K + 1 classes) for each selected biomarker is employed to build the classification. Since a grouped-lasso penalty is imposed on each biomarker, the effects of a biomarker over the outcome classes will all be zero or nonzero. The approach can be executed using the R package “glmnet.”
In the machine learning (ML) framework, we first utilize unsupervised learning feature selection to pick the top
Results
Simulation studies: Synthetic dataset with complex gene structure
We are currently conducting a numerical analysis to showcase our proposed OGS method with multinomial logistic regression. Additionally, we aim to evaluate the predictive efficacy of our method in comparison to several established machine learning approaches. Synthetic data consisting of 500 samples are utilized as the training set, with each subject’s responses generated from a 3-class multinomial distribution,

with the covariates

where
The simulation considers the gene group size (the number of genes per group) and the overlapping structure (the number of genes shared by two overlapping groups), as outlined in Table 1, where we can see, for instance, groups 10 and 11 each consist of 15 genes, totaling 25 unique genes between them, with 5 genes shared. Overall, this study encompasses 193 genes and 243 potential group-specific gene effects. Figure 1 illustrates the associated gene network structure. We further hypothesize that different biomarker effects are present in different outcome classes. In class 1, we hypothesize the efficacy of gene groups 9 and 11, with genes in each group exhibiting consistent effects of −1.5 and 1.5, respectively. Moreover, within group 9, effective G-G interactions (G37-G39, G38-G40) demonstrate effects of (1.5, 1.5), while between groups 9 and 11, effective G-G interactions (G41-G58, G42-G59) display effects of (1.5, 1.5). In class 2, we hypothesize the efficacy of gene groups 13 and 15, with genes in each group exhibiting consistent effects of 1.5 and 1.5, respectively. Moreover, within group 13, effective G-G interactions (G78-G80, G79-G81) demonstrate effects of (1.5, 1.5), while between groups 13 and 15, effective G-G interactions (G82-G118, G83-G119) display effects of (1.5, 1.5). There are 18 721 major genes and G-G interaction pairs in this simulation study, and the average proportions of outcome classes 1, 2, and 0 are 35%, 39%, and 26%, respectively.
The gene group structure for the varying gene group-size data.


The gene network structure for the varying gene group-size data.
We conducted the described simulation setup 500 times to gather numerical results. The results presented in Table 2 indicate that the OGS method employing Ridge, Lasso, and Adaptive Lasso penalties consistently outperforms other methods, including common ML techniques, in multi-class prediction.
Averages (standard deviations) of testing prediction performance over 500 simulated replicates for various multi-class classification methods under the gene structure with different gene group sizes.

We also explore an alternate gene network structure, comprising 18 groups, each containing 10 genes. Details regarding group sizes and overlapping structure are provided in Table 3. This example encompasses 129 genes and 180 potential group-specific gene effects. Figure 2 illustrates the associated gene network structure. In class 1, we hypothesize the efficacy of gene groups 1 and 4, with genes in each group exhibiting consistent effects of −1.5 and 1.5, respectively. Moreover, within group 1, effective G-G interactions (G1-G3, G2-G4) demonstrate effects of (1.5, 1.5), while between groups 1 and 4, effective G-G interactions (G5-G25, G6-G26) display effects of (1.5, 1.5). In class 2, we hypothesize the efficacy of gene groups 13 and 18, with genes in each group exhibiting consistent effects of 1.5 and 1.5, respectively. Moreover, within group 18, effective G-G interactions (G123-G125, G124-G126) demonstrate effects of (1.5, 1.5), while between groups 13 to 18, effective G-G interactions (G88-G127, G89-G128) display effects of (1.5, 1.5). There are 8385 major genes and G-G interaction pairs in this simulation study, and the average proportions of outcome classes 1, 2, and 0 are 38%, 36%, and 26%, respectively.
The gene group structure for the equal gene group-size data.


The gene network structure for the equal gene group-size data.
From the results presented in Tables 2 and 4, it’s apparent that the OGS method with Ridge, Lasso, and Adaptive Lasso penalties consistently outperforms other methods, including traditional ML approaches, in terms of classification performance. Additionally, both Tables 2 and 4 showcase the standard deviations of accuracy metrics across different methods, indicating that the OGS methods exhibit slightly higher variability in accuracy compared to alternative approaches.
Averages (standard deviations) of testing prediction performance over 500 simulated replicates for various multi-class classification methods under the gene structure with equal gene group sizes.

Real data application: Kaplan-Meier survival curves
We first display the 3 Kaplan-Meier survival curves for the 3 cancer types (KIRP, LUAD, and HNSCC) across the 3 groups (alive, dead with no tumor, and dead with tumor). We then perform a log-rank test to assess whether there are significant differences between the survival curves of these 3 groups. From Figure 3, it can be observed that there are significant differences in the survival curves among the 3 groups in the survival data of KIRP and HNSCC. However, in the survival data of LUAD, there are no significant differences in the survival curves between the “dead with no tumor” and “dead with tumor” groups.

Kaplan-Meier survival outcomes for the three cancer types (KIRP, LUAD, and HNSCC) across the three groups (alive, dead with no tumor, and dead with tumor).
Real data application: TCGA KIRP data
Our own TCGA KIRP data consist of 275 subjects, of whom 235 (85.5%) alive, 12 (4.4%) dead with no tumor, and 28 (10.0%) dead with tumor. The data is extremely imbalanced in terms of the outcome class distribution. Given that the pool of cancer-related genes is likely finite, it makes sense to streamline the gene set before constructing the classification model. We employ unsupervised learning for feature selection, identifying the top 1000 genes with the most significant absolute variation for subsequent analysis.
For the proposed OGS approach, out of the initial 1000 genes selected through unsupervised learning, 697 genes are linked to 398 pathways based on prior pathway information from the GO Cellular Component (GO-CC) database. The remaining 303 genes, not mapped to any pathway in the GO-CC database, are either excluded or grouped together in the OGS method. These alternative approaches result in a total of 243 253 and 500 500 main and G-G interaction effects, respectively.
We randomly split the entire dataset into 10 sets of 165:110 for 60% training and 40% testing, respectively, to evaluate the performance of all considered methods. The ROSE resampling is performed on the training data to address the class imbalance issue. Table 5 summarizes the average 10-fold classification results after removing 303 ungrouped genes from the analysis. We also consider another pathway database, Kyoto Encyclopedia of Genes and Genomes (KEGG),35 -37 and the corresponding analysis results are shown in Table A.2 of Appendix. From both sets of results, we see that the OGS method has better classification performance compared to the other methods in terms of REC, PRE, and F performance metrics. The ML methods SVM and KNN have superior performance in terms of the metric OA but inferior REC, PRE, and F metrics compared to the OGS, owing to that the ML methods perform well in the majority outcome classes (alive and dead with tumor outcomes), but perform poorly in the minority class (dead with no tumor outcome).
Averages (standard deviations) of testing prediction performance of different methods with GO_CC gene sets databases in the TCGA KIRP data over 10 random splits of 165:110 training/test sets.

Next, based on the GO-CC database, we apply the OGS approach with the adaptive lasso penalty to the entire TCGA KIRP data, and examine the selected features in the dead with tumor outcome category. The method selects 95 G-G interaction biomarkers, and the corresponding network is shown in Figure 4. Some selected biomarkers have been shown to have biological meaningful in published literature. For example, Wang et al 38 showed that the “HOXDs” gene is lowly expressed in KIRP, and the upregulation of “HOXDs” is associated with improved overall survival of cancer patients. These findings suggested that “HOXDs” may be an indicator biomarker for pan-cancer prognosis and immunotherapy. Jia et al 39 demonstrated the expression and function of “CAMK2B” in vitro and in vivo, and provided evidence that this protein promotes reregulation of the stromal tumor microenvironment and inhibits KIRP proliferation.

The network of the selected G-G interactions by the OGS approach with the adaptive lasso penalty in the TCGA KIRP gene expression data with dead with tumor outcome.
Real data application: TCGA LUAD data
The TCGA LUAD data consist of 454 subjects, of whom 304 (67.0%) alive, 38 (8.4%) dead with no tumor, and 112 (24.7%) dead with tumor. There exists class imbalance in this dataset. We choose the top 1000 genes with the highest absolute variation for subsequent analysis.
For the proposed OGS approach, out of the initial 1000 genes selected through unsupervised learning, 640 genes are linked to 402 pathways based on prior pathway information from the GO-CC database. The remaining 360 genes, not mapped to any pathway in the GO-CC database, are either excluded or grouped together in the OGS method. These alternative approaches result in a total of 205 120 and 500 500 main and G-G interaction effects, respectively. We randomly split the entire dataset into 10 sets of 272:182 for 60% training and 40% testing, respectively, to evaluate the performance of all considered methods. The ROSE resampling is performed on the training data to alleviate the class imbalance.
Table 6 summarizes the average 10-fold classification results after removing 360 ungrouped genes from the analysis. We also consider KEGG pathway database, and the corresponding analysis results are shown in Table A.3 of Appendix. From both sets of results, it is apparent that the OGS method consistently exhibits superior classification performance in terms of REC, PRE, and F metrics compared to other methods. The SVM and KNN have superior performance in terms of the metric OA but inferior REC, PRE, and F metrics compared to the OGS, owing to that the ML methods perform well in the majority outcome classes (alive and dead with tumor outcomes), but perform poorly in the minority class (dead with no tumor outcome).
Averages (standard deviations) of testing prediction performance of different methods with GO_CC gene sets databases in the TCGA LUAD data over 10 random splits of 272:182 training/test sets.

Based on the GO-CC database, the OGS approach with the adaptive lasso penalty selects 121 G-G interaction biomarkers, and the corresponding network is shown in Figure 5. Some selected biomarkers have been shown to have biological meaningful in published literature. For example, Zhang et al 40 showed that the gene “JPH3” was associated with non-small cell lung cancer (NSCLC), and they found that some genes including “JPH3” were frequently silenced by epigenetic mechanisms in lung cancer. Also, Nasser et al 41 demonstrated that “S100A7” is upregulated in multiple types of malignancies, including non-small cell lung cancer, contributing to tumor growth, premetastatic niche formation, and metastasis.

The network of the selected G-G interactions by the OGS approach with the adaptive lasso penalty in the TCGA LUAD gene expression data.
Real data application: TCGA HNSCC data
The TCGA HNSCC data consist of 491 subjects, of whom 296 (60.3%) alive, 64 (13.0%) dead with no tumor, and 131 (26.7%) dead with tumor. The data is moderately imbalanced in terms of the outcome class distribution. We choose the top 1000 genes with the highest absolute variation for subsequent analysis.
For the proposed OGS approach, out of the initial 1000 genes selected through unsupervised learning, 667 genes are linked to 393 pathways based on prior pathway information from the GO-CC database. The remaining 333 genes, not mapped to any pathway in the GO-CC database, are either excluded or grouped together in the OGS method. These alternative approaches result in a total of 222 778 and 500 500 main and G-G interaction effects, respectively. We randomly split the entire dataset into 10 sets of 295:196 for 60% training and 40% testing, respectively, to evaluate the performance of all considered methods. The ROSE resampling is performed on the training data to alleviate the class imbalance.
Table 7 summarizes the average 10-fold classification results after removing 333 ungrouped genes from the analysis. We also consider KEGG pathway database, and the corresponding analysis results are shown in Table A.4 of Appendix. These results reveal that, the OGS approach has slightly better performance metrics than the ML methods in terms of REC, PRE, and F classification metrics, which focus more on the rare class, while the ML method SVM has the best classification performance in terms of the OA metric, which focuses more on the dominant classes.
Averages (standard deviations) of testing prediction performance of different methods with GO_CC gene sets databases in the TCGA HNSCC data over 10 random splits of 295:196 training/test sets.

Based on the GO-CC database, the OGS approach with the adaptive lasso penalty selects 85 G-G interaction biomarkers, and the corresponding network is shown in Figure 6. Some selected biomarkers have been shown to have biological meaningful in published literature. For example, Irimie et al 42 showed that the gene “MGST1” was associated with HNSCC, and they found that the expression levels of several genes, including the “MGST1” gene, were altered between smoking and nonsmoking HNSCC patients. Misawa et al 43 showed that neuropeptide genes including “GAL” are powerful epigenetic biomarkers in HNSCC.

The network of the selected G-G interactions by the OGS approach with the adaptive lasso penalty in the TCGA HNSCC gene expression data.
We also report the biomarkers with the top- and bottom- coefficients for the “dead with tumor” outcome, identified using the OGS approach with adaptive lasso penalty to the 3 entire TCGA transcriptomic data, based on the GO-CC and KEGG databases. The results are detailed in Table 8 and A.5 of Appendix. Positive coefficients indicate that higher biomarker expression increases “dead with tumor” event probability, while negative coefficients indicate it decreases event probability.
Biomarkers with the top- and bottom-coefficients for the “dead with tumor” outcome are identified using the OGS approach with the adaptive lasso, based on GO-CCdatabase.

Additionally, we plot the receiver operating characteristic (ROC) curves for the “dead with tumor” category of these 3 real datasets using various classification methods, and calculated the corresponding area under curve (AUC) values for both training and testing data. The ROC curves and AUC values for all methods are obtained by averaging the results from 10 iterations of the validation set approach. The corresponding graphs are shown in Figure 7 and A.1 of Appendix. From the ROC curves and AUC values in these 2 figures, we can conclude that our proposed method effectively avoids the overfitting problem compared to the common machine learning methods considered in the article.

Average ROC curves and AUC values for “dead with tumor” across KIRP, LUAD, and HNSCC datasets, evaluated with different classification methods on training and test data, based on GO-CC database.
The advantage of our proposed method compared to machine learning approaches is that we emphasize model inference. Specifically, we focus on understanding the relationship between important biomarkers and the response variable, not black box models. In terms of prediction, our method allows us to calculate the probability that an observation belongs to a certain class, rather than merely predicting a classification. ROC curve analysis of real data also indicates that our method can avoid overfitting, which is a common issue with machine learning methods.
Discussion
In summary, we outline the similarities and distinctions among Wang et al, 17 Wang and Chen, 4 and the current paper. Wang et al 17 employed the OGS approach with Cox’s regression model to identify significant gene-environment interactions linked to clinical censoring survival outcomes. Conversely, Wang and Chen 4 utilized the OGS approach with a binary logistic regression model to discover critical gene-gene interaction biomarkers associated with the occurrence of binary cancer/normal outcomes. This article employs the OvO strategy to convert multi-class classification into multiple binary classifications. It then integrates this approach with the OGS procedure, utilizing a binary logistic regression model as outlined by Wang and Chen. 4 This combination aims to identify significant gene-gene interaction biomarkers associated with multiple survival statuses in cancer patients. In this paper, in addition to the typical challenges of ultra-high dimensionality and feature contamination, we also encounter the problem of data imbalance. Together, these factors pose significant obstacles to accurate predictive modeling.
Potential improvements to the OGS method
Since the real-world data we are interested in is imbalanced, there are 3 main approaches to dealing with class imbalance: resampling, cost-sensitive, and ensembling, and several extensions based on these approaches have also been developed.19,44 -46 In the real data applications, we just leverage the ROSE resampling procedure to balance the data, while a remaining interesting problem is, how to find a best way to tackle class imbalance for downstream genome-wide association study (GWAS), and how this way may work with the OGS approach.
In practical data analysis, we first select the top 1000 genes with the highest variance in gene expression. However, since variance itself is susceptible to outliers and gene data is often contaminated, it is essential to explore more suitable unsupervised feature selection methods. 47 Moreover, Fan and Lv 15 pointed out marginal feature selection may overlook key predictors due to: (1) Joint correlation not captured by marginal analysis, (2) Selection of secondary predictors highly correlated with important ones, and (3) Collinearity among predictors. They proposed an iterative method to address these issues. In addition, feature selection is the process of trying to select more informative features. Too many redundant or irrelevant features may overwhelm the important features of the classification. Feature selection can solve such problems, thereby improving prediction accuracy and reducing the computational cost of classification algorithms. Another interesting issue is that after the OGS procedure selects the most important genes and gene pairs, we can try feeding these selected biomarkers into another machine learning algorithm to see how well the predictions perform. We will study these further issues in future work.
In the OGS method, the SKAT test is key for screening gene interactions. Lee et al 48 evaluated various gene- or region-based testing methods, including burden and variance-component tests, and assessed their performance. Since different methods have unique strengths based on the biological context, future research should explore diverse testing approaches to enhance OGS effectiveness. The OGS method extracts gene network information using predefined pathways, which limits it to genes in those pathways and can result in information loss. Researching ways to relax these constraints could improve feature selection and classification prediction. Besides, we utilize two-way and multiplicative interactions for simplicity in interaction assessments. However, higher-order and more complex interactions are challenging and warrant further research.
Misclassification analysis discussing
In this study, based on the classification by Feng et al, 9 we divided cancer patients into 3 groups according to their survival status and clinical condition (ie, dead due to cancer, dead due to other reasons, and alive). However, this classification could be further refined, for example: (1) Survivors Close to Death: If some survivors are in a health condition very close to death, this might impact the accuracy and interpretation of the classification results. Introducing indicators of health severity could improve classification accuracy. (2) Death without Tumor Population: If some individuals may not have been diagnosed with tumors or if the cause of death records are inaccurate, this could affect the accuracy of the analysis. To mitigate this impact, using grading or other indicators to assess the actual condition of these patients could be considered. (3) Censoring Issues: In survival data, censoring is an important consideration. Future research should incorporate censoring information into the cancer patient classification to achieve more accurate classification and prediction. These considerations could help improve the precision of the classification and the reliability of the analysis, leading to a better understanding of cancer patients’ survival conditions and treatment outcomes.
Multiple cancer subtypes classification
van’t Veer and Bernards 49 highlighted that identifying cancer subtypes is crucial for personalized precision medicine, as treatment decisions heavily depend on understanding these subtypes. For instance, Lavagna et al 3 developed a biomarker predictor to classify small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), while Tian et al 50 used a network-constrained sparse multinomial logit model to predict glioblastoma multiforme (GBM) subtypes. Tabibu et al 51 applied deep learning to classify renal cell carcinoma and predict survival outcomes based on pathological images. Such classification methods support early diagnosis, enabling more precise treatments and prognostic assessments. Consequently, the OGS with multinomial logistic regression model could also be used for cancer subtype identification and prediction.
State-of-the-art methods
Recent studies at the forefront of the field have demonstrated improved classification results. It is widely recognized that metaheuristic algorithms have been extensively utilized to enhance classification performance. For example, the hybridization of Particle Swarm Optimization has improved crime rate prediction. 52 Similarly, combining Cuckoo Search with Harris Hawks Optimization has boosted cancer detection rates, 53 while integrating Cuckoo Search with deep learning has enhanced cancer disease classification. 54 Additionally, Marine Predator Chaotic Search has proven effective for detecting COVID-19. 55 Therefore, we are going to conduct a series of investigations and studies to examine the performance of different state-of-the-art methods on multiclass imbalanced biological data.
Large datasets integrations
Several public human databases, including Gene Expression Omnibus (GEO), Molecular Taxonomy of Breast Cancer International Consortium (METABRIC), National Cancer Database (NCDB), and The Cancer Genome Atlas (TCGA), are valuable for assessing the reproducibility of our findings. We believe that conducting a meta-analysis could help discover and validate survival prognostic biomarkers 56 and plan to explore this in future research.
Conclusion
In this article, we employ the OvO strategy to transform multi-class classification into multiple binary classification, and utilize the OGS with binary logistic regression to include gene pathway information for identifying important major genes and gene-gene interactions for multicategory survival outcomes. Based on the identified biomarkers, we can predict for each patient the probabilities that he/she belongs to each of the outcome classes. In simulation studies, we demonstrate that the classification performance of our proposed method outperforms some commonly used ML methods and the multinomial logistic regression with the group lasso penalty. In real data applications, we employ the ROSE resampling procedure to address the class imbalance and analyze 3 sets of TCGA cancer transcriptomic data (KIRP, LUAD, and HNSCC). The numerical results demonstrate that the new proposal leads to a substantial improvement in cancer diagnosis when compared to methods that do not take pathway information into account.
Supplemental Material
sj-docx-1-cix-10.1177_11769351241286710 – Supplemental material for Multicategory Survival Outcomes Classification via Overlapping Group Screening Process Based on Multinomial Logistic Regression Model With Application to TCGA Transcriptomic Data
Supplemental material, sj-docx-1-cix-10.1177_11769351241286710 for Multicategory Survival Outcomes Classification via Overlapping Group Screening Process Based on Multinomial Logistic Regression Model With Application to TCGA Transcriptomic Data by Jie-Huei Wang, Po-Lin Hou and Yi-Hau Chen in Cancer Informatics
Footnotes
Acknowledgements
Author Contributions
JH conceived and designed the experiments. PL collected and organized the analysis data. JH and PL analyzed the data. JH and YH wrote the first draft of the manuscript. JH and YH made critical revisions and approved final version. All authors agreed with manuscript results and conclusions. All authors jointly developed the structure and arguments for the paper. All authors reviewed and approved of the final manuscript.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the grant NSTC 112-2118-M-194-003-MY2 from the National Science and Technology Council of Republic of China (Taiwan). The funding body did not play any role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of Data and Materials
The R codes for both simulation studies and real data applications can be accessed on the figshare website: https://doi.org/10.6084/m9.figshare.23849679.v2. Additionally, the transcriptomic data for TCGA KIRP, LUAD, and HNSCC, along with the clinical multicategory survival outcomes examined in this study, are available on figshare: https://doi.org/10.6084/m9.figshare.23849370.v3. The TCGA data analyzed in this study were originally obtained from the TCGA Hub repository: https://tcga.xenahubs.net, with the primary source being the TCGA Website: ![]() .
.
Ethics Approval and Consent to Participate
The study described in this manuscript did not include human or animal participants. All data used in this research were sourced from publicly available and freely accessible repositories. Therefore, ethical approval was unnecessary for this study.
Consent for Publication
Not applicable.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.