
Abstract
Objective:
The aim of this study was to evaluate the post-marketing safety, tolerability, immunogenicity and efficacy of Bevacizumab (manufactured by Hetero Biopharma) in a broader population of patients with solid tumors.
Patients And Methods:
This phase IV, prospective, multi-centric clinical study was carried out in Indian patients with solid malignancies (metastatic colorectal cancer, non-squamous non-small-cell lung cancer, metastatic renal cell carcinoma) treated with Bevacizumab between April 2018 and July 2019. This study included 203 patients from 16 tertiary care oncology centers across India for safety assessment, of which a subset of 115 patients who have consented were also evaluated for efficacy and immunogenicity. This study was prospectively registered in the Clinical Trial Registry of India (CTRI), and was commenced only after receiving approval from the competent authority (Central Drugs Standard Control Organization, CDSCO).
Results:
Out of the 203 enrolled patients, 121 (59.6%) patients reported 338 adverse events (AEs) during this study. Of 338 reported AEs, 14 serious adverse events (SAEs) were reported by 13 patients including 6 fatal SAEs, assessed as unrelated to the study medication and 7 non-fatal SAEs, 5 assessed as related, and 3 unrelated to Bevacizumab. Most AEs reported in this study (33.9%) were general disorders and administration site conditions, followed by gastrointestinal disorders (29.1%). The most frequently reported AEs were diarrhea (11.3%), asthenia (10.3%), headache (8.9%), pain (7.4%), vomiting (7.9%), and neutropenia (5.9%). At the end of the study, 2 (1.75%) of 69 patients reported antibodies to Bevacizumab without affecting safety and efficacy. However, at the end of 12 months, no patient had reported antibodies to Bevacizumab. Complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD) were reported in 18.3%, 22.6%, 9.6%, and 8.7% of patients, respectively. The overall response rate (CR + PR) was reported in 40.9% of patients at the end of the study. Disease control rate (DCR), also known as the clinical benefit rate (CBR) was reported in 50.4% of patients.
Conclusions:
Bevacizumab (Cizumab, Hetero Biopharma) was observed to be safe, well tolerated, lacking immunogenicity, and efficacious in the treatment of solid tumors. The findings of this phase IV study of Bevacizumab, primarily as a combination therapy regimen suggest its suitability and rationality for usage in multiple solid malignancies.
Clinical Trial Registry Number:
CTRI/2018/4/13371 [Registered on CTRI http://ctri.nic.in/Clinicaltrials/advsearch.php : 19/04/2018]; Trial Registered Prospectively.
Keywords
Introduction
Bevacizumab, a recombinant humanized vascular endothelial growth factor (VEGF) monoclonal antibody (mAb), was approved by the US Food and Drug Administration (FDA) on February 2004. 1 It binds and neutralizes all isoforms of VEGF and prevents VEGF from binding to its receptors (VEGFR-1 and VEGFR-2), thereby inhibiting angiogenesis and tumor growth, normalization of remaining tumor vasculature, and inhibition of further tumor angiogenesis reflecting a key targeting strategy in cancer therapy. 2 Bevacizumab remodels existing tumor vasculature by decreasing vessel diameter, density, and permeability. 3 Bevacizumab was approved initially for the treatment of metastatic colorectal cancer (mCRC) and subsequently was approved for the treatment of multiple recurrent or metastatic non-squamous non-small-cell lung cancer (NSCLC), metastatic renal cell carcinoma (mRCC), metastatic cervical cancer, recurrent glioblastoma (GBM), hepatocellular carcinoma (HCC) epithelial ovarian, fallopian tube, and primary peritoneal cancers by the FDA 4 in the United States.
Colorectal cancer (CRC) remains a major public health problem in most developed countries. In 2008, a global incidence of about 1.2 million was reported. Based on currently available data, in India the CRC incidence rates for males and females are 4.3 per 100 000 population and 3.4 per 100 000 population, respectively. While the incidence rate of CRC among native Indians has been slowly rising over many decades, the incidence of CRC in immigrant Indians living in the UK and USA has raised rapidly. In India, the absolute burden of CRC has also increased over the past 3 decades. 5 NSCLC accounts for over 85% of lung cancer diagnoses, the leading cause of cancer-related death worldwide, and most patients present with advanced stage at the time of diagnosis, resulting in a poor prognosis. Currently, results of large-scale randomized trials and real-world studies have placed Bevacizumab in combination with platinum-based chemotherapy as the standard first-line therapy for non-squamous (NS)-NSCLC. 6 However, the cost of Bevacizumab was unaffordable for the majority of the population in the developing countries. Hence, Hetero has developed a biosimilar of Bevacizumab to reduce the cost and reach the needy at an affordable cost. Hetero conducted a phase III clinical trial to evaluate the efficacy, safety, and immunogenicity in mCRC patients. In our phase III study, disease control rate (DCR) of 96.97%, 98.04%, 89.80% and overall response rate (ORR) of 46.97%, 49.02%, 46.94% were reported at 12, 18, and 24 weeks, respectively. Anti-Bevacizumab antibodies were reported in 4 (9.76%) patients at the end of 12 weeks, without affecting safety and efficacy. Post phase III study, Hetero received manufacturing and marketing authorization for mCRC, NSCLC, GBM, mRCC, persistent, recurrent, or metastatic carcinoma of the cervix, and mBC.
As per the Guidelines for Similar Biologics (2016), India, 16 in order to further reduce the residual risk of the Similar Biologics, additional safety data may need to be collected after market approval through a pre-defined single arm study of generally more than 200 evaluable patients and compared to historical data of the Reference Biologic. The primary aim of the post-marketing phase IV study is safety and secondary endpoint shall include efficacy and immunogenicity. In line with the regulatory requirements, this phase IV post-marketing study was conducted in patients with all approved indications in 16 oncology tertiary care centers in India, to evaluate long term safety, immunogenicity, and efficacy.
Material and Methods
Study design
This was a phase IV, prospective, open-label, multi-center, post-marketing study of the marketed formulation of Hetero-Bevacizumab (Cizumab) in patients with solid malignancies, conducted between April 2018 and July 2019 at 16 sites in India to evaluate the safety, efficacy, and long-term immunogenicity of Bevacizumab in real-life scenario. This phase IV study was approved by Central Licensing Authority of India (CDSCO) on 13/05/2016, and the same was prospectively registered with CTRI on 19/04/2018 (CTRI/2018/4/13371). Trial approval was obtained from all independent ethics committees of the participating institutes before it was conducted.
Sample size
This study aimed to collect data on at least 200 patients with solid malignancies, in compliance with Similar Biologics Guidelines, India in order to monitor and report long-term safety, immunogenicity, and efficacy. Assuming loss to follow up, 214 patients were screened to enroll 203 patients. All 203 patients were included in the safety evaluation and 115 patients who had consented for additional assessments, evaluated for immunogenicity and efficacy.
Inclusion and exclusion criteria
The study included male or female adult patients of 18 years or older, with solid malignancies (mCRC, NSCLC, GBM, mRCC, mBC and persistent, recurrent, or metastatic carcinoma of the cervix), patient with low risk for obstruction or perforation, with adequate hematological function that is, absolute Neutrophil count (ANC) ⩾1.5 × 109/L; platelet count ⩾100 × 109/L, Hemoglobin (Hb) ⩾9 g/dL, major surgery, open surgical biopsy, or significant traumatic injury more than 4 weeks before randomization. Patients with serious hemorrhage or recent hemoptysis; presence of a serious, non-healing wound, ulcer, or bone fracture; history of hemorrhagic diathesis or coagulopathy; significant vascular disease (eg, aortic aneurysm or dissection) or recent peripheral arterial thrombosis within 6 months prior to dosing; any peripheral vascular disease; or venous thrombo-embolic events within the past 3 months; planned pregnancy or lactating; clinical history/evidence of allergy/hypersensitivity to murine proteins; history or pre-existing clinical evidence of medically significant abnormality or requirements for surgery were excluded from the study.
Hetero-Bevacizumab was administered in the dosage and for the duration, as recommended in the package insert (Table 1). In the event of gastrointestinal perforations, fistulas involving internal organs, wound dehiscence and wound healing complications requiring medical intervention, serious hemorrhage, severe arterial or venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome (PRES), or nephrotic syndrome, the study drug was discontinued.
Dosage and administration of Bevacizumab in solid tumors.

Study assessments
Safety assessments were conducted on all patients at screening, end of treatment up to 6 to 8 cycles or end of study. A subset of 115 patients were also assessed for long term immunogenicity and efficacy that is, at screening, at the end of 6 to 8 cycles, or at the end of the study. During study treatment, all patients were monitored for significant changes in clinical signs and symptoms, laboratory abnormalities. We assessed immunogenicity was assessed at baseline and at the end of the 12-month period.
Treatment emergent clinical & laboratory adverse events (TEAEs) were assessed as a primary endpoint. An adverse event was defined as occurrence of any untoward medical emergency in a patient, who had been administered a medicinal product and which occurrence does not necessarily have to have a causal relationship with this treatment. A serious adverse event was defined as any adverse event associated with death, inpatient hospitalization (in case the study was being conducted on out-patients), prolongation of hospitalization (in case the study was being conducted on in-patients), persistent or significant disability or incapacity, a congenital anomaly or birth defect, or is otherwise life-threatening. 7 Adverse events were evaluated for their causality, expectedness, seriousness, incidence, severity, outcome, duration, and action taken. Immunogenicity was evaluated by assessing the presence of anti-Bevacizumab antibodies in serum by using a validated bioanalytical method at Hetero Biopharma Limited. The immunogenicity assessment for the anti-Bevacizumab antibody is performed by the Affinity capture and elution (ACE) method by enzyme-linked immunosorbent assay (ELISA). The method follows sequential steps to detect anti-drug antibody (ADA) in the presence of high levels of free drug (Bevacizumab). First, the ADA-free drug complex was dissociated with acid treatment followed by neutralization in the presence of solid–phase drug giving the ADA an opportunity to be affinity captured. After washing away excess free drug, bound ADA were eluted with acid and subsequently bound to a fresh solid surface upon neutralization. Bound ADA was subsequently detected by biotin labeled drug and Streptavidin Horseradish Peroxidase (HRP). Tetramethylbenzidine (TMB) substrate was used to generate a response. The Optical Density (OD) of the wells was measured using an ELISA plate reader and it was directly proportional to the amount of anti-Bevacizumab antibody present in the sample. 8 Efficacy assessments in terms of ORR and DCR were assessed by the central radiology team according to RECIST 1.1 criteria. 9 Additionally, immunogenic reactions and an improvement in response rates were evaluated as potential reasons for lack of efficacy.
Statistical analysis
All patients who have taken at least one dose of study drug were included in safety population, all patients who completed at least one dose of study drug and have one post baseline efficacy assessments were included in the intent to treat (ITT) population, and patients who completed the study treatment as per the protocol without any major protocol deviations were included in per protocol (PP) population. The continuous variables like age, body weight, body surface area, height, number of target lesions were summarized by number (N), mean, median, standard deviation, and range (minimum, maximum). All the categorical variables like gender were summarized with counts and percentages. Efficacy endpoints at baseline and subsequent visits were analyzed using Logistic Regression (or) Fisher’s exact test. The 95% confidence interval for the mean change was also estimated. All statistical tests were performed using two-tailed tests at a 5% level of significance. Safety variables including AEs, clinical laboratory parameters, vital signs, ECG parameters, and complete physical examinations were listed and summarized with descriptive statistics as appropriate. AEs were coded using version 23.0 of the Medical Dictionary for Regulatory Activities (MedDRA). 10 All statistical tests were performed using SAS® (version 9.4 or higher) system software (SAS Institute Inc.; USA).
Ethical approval
This study was conducted in accordance with the ethical guidelines set out in the Declaration of Helsinki, 1964 (revised in 2013), Post Authorization Safety Studies (phase IV study), Schedule Y, Drug & Cosmetic Act 201, and Guidelines for Similar Biologics 2016, India, along with subsequent amendments and Indian regulatory laws governing biomedical research involving human patients. Institutional Ethics Committee approvals were obtained from each participating study center before initiating the study and written informed consent was obtained from each study participant before any protocol-driven tests or evaluations were performed.
Results
Patient characteristics
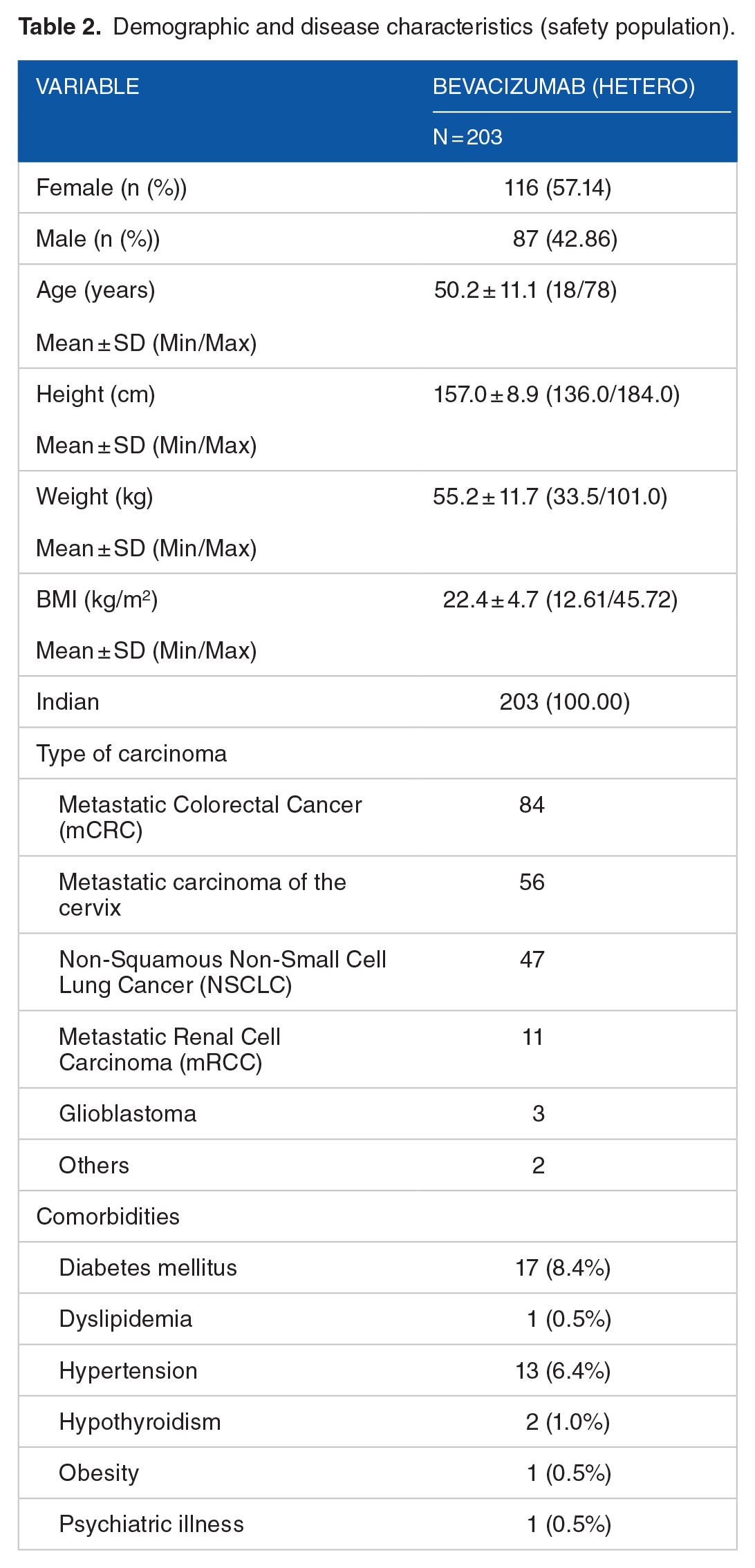
A total of 214 patients were screened to enroll 203 patients in this study. Out of 203 patients enrolled, 115 patients who had consented for additional assessments, were evaluated for efficacy. Female patients were more with 57.14% compared to 42.86% male patients. The average age of the group included was 50.2 years old, ranging from 18 to 78 (Table 2). Fifty-six patients out of 115 who were evaluated for efficacy, completed the study (please refer to indication-wise disposition in Table 3 and Figure 1). Overall, 84 patients of mCRC, 56 patients of mCaCx, 47 patients of NSCLC, 11 patients of mRCC, 3 patients of Glioblastoma, and 2 patients of other gastrointestinal and peritoneal cancer were included in this study.
Demographic and disease characteristics (safety population).
Indication-wise patient disposition in solid tumors.

Abbreviations: mCC, metastatic carcinoma of the cervix; mCRC, metastatic colorectal cancer; mRCC, metastatic renal cell carcinoma; NS-NSCLC, non-squamous non-small-cell lung cancer.

Indication-wise patient disposition.
Safety evaluation
On an average, all patients were given 4 to 8 cycles of treatment over a period of 11 to 15 weeks. Overall, 121 patients (59.6%) reported one or more adverse events during the study period (Table 4). Out of 121 patients, 69 (33.9%) patients reported 105 (31.1%) AEs in general disorders and administration site conditions, 59 (29.1%) patients reported 63 (18.6%) AEs in gastrointestinal disorders, 36 (17.7%) patients reported 54 (15.9%) AEs in blood and lymphatic system disorders, 26 (12.8%) patients reported 28 (8.3%) AEs in nervous system disorders, and 21 (10.3%) patients reported 24 (7.1%) AEs in investigations (Table 3). The most frequently reported AEs were diarrhea, asthenia, headache, pain, neutropenia, and vomiting. Most AEs were mild to moderate in intensity. Seventy-seven (22.8%) AEs were considered as related, and 261 (77.2%) AEs were not related to the study drug as assessed by the principal investigator. Out of the 338 AEs, 14 SAEs were reported by 13 patients. Of these SAEs, 6 deaths were reported that were not related to study medication and all remaining SAEs were resolved.
List of adverse events occurred in patients with solid tumors treated with Bevacizumab.
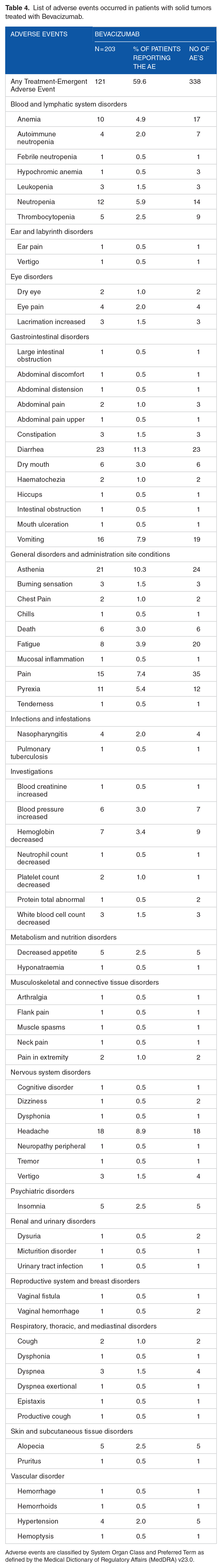
Adverse events are classified by System Organ Class and Preferred Term as defined by the Medical Dictionary of Regulatory Affairs (MedDRA) v23.0.
Immunogenicity analysis
Immunogenicity was assessed for 114 patients who had baseline negative antibodies. Out of 114 patients, 69 patients completed the treatment with 2 (2.9%) patients developed anti-Bevacizumab antibodies. Two patients who had anti-Bevacizumab antibodies at the end of study, did not report any safety concerns during follow-up and it did not affect the overall efficacy. Further, only 22 patients could provide 12 months immunogenicity samples and no patients had anti-Bevacizumab antibodies (Table 5).
Summary of immunogenicity assessment.

End of the study: At the end of 6 to 8 cycles of treatment or at study completion whichever is earlier.
End of 12 months: At the end of 12-month from start of study treatment.
Efficacy analysis
Overall, a total of 21 (18.3%), 26 (22.6%), 11(9.6%), and 10 (8.7%) patients reported CR, PR, SD, and PD, respectively, in ITT population (n = 115). Overall response rate (ORR) was reported in 17 (40.9%) patients at the end of the study. The DCR was reported in 58 (50.4%) patients. Tumor response data was not available for analysis in 47 (40.9%) patients. In the PP population (n = 56), CR, PR, SD, and PD were reported in 18 (32.1%), 23 (41.1%), 9 (16.1%), and 6 (10.7%) patients, respectively. The ORR was reported in 41 (73.21%) patients and DCR was reported in 50 (89.29%) patients at the end of the study. The tumor response rate at EOS has been presented in Table 6.
Summary of tumor response rate at EOS – ITT population.

Discussion
Biologics have revolutionized the treatment of many serious diseases, including cancer, rheumatoid arthritis, and inflammatory bowel disease (IBD); however, the issue of immunogenicity (ie, the development of anti-drug antibodies (ADA) against these protein-based therapies) continues to concern patients and health care providers. Long-term outcomes of diseases treated with biosimilars may be severely impacted by immune responses to them, necessitating increased vigilance against ADA formation and the consequent loss of treatment response to the few agents approved in this vulnerable patient population.
Bevacizumab (manufactured by Hetero under the brand name Cizumab) is a recombinant, humanized anti-VEGF monoclonal antibody that inhibits VEGF function in vascular endothelial cells and thereby inhibits tumor angiogenesis, upon which solid tumors depend for growth and metastasis.
As per the Similar Biologics Guidelines, CDSCO, India, this study was a non-comparative single arm study to assess the post-marketing long term safety, immunogenicity, and efficacy of Hetero’s Bevacizumab in solid tumors. Possibility of assessment bias cannot be excluded due to single arm and non-comparative nature of this study. This was a post-marketing study conducted at various oncology tertiary care centers without any strict inclusion and exclusion criteria resulting in possible confounding factors like concomitant diseases, medication, and other disease related factors, which might affect the study outcomes. There was no formal sample size calculation to evaluate significant differences in the study outcomes. However, the study data was analyzed and presented in terms of descriptive statistics.
Historically, the most important adverse events associated with Bevacizumab include bleeding/hemorrhage (39.1%), pulmonary hemorrhage (2.1%), proteinuria (10.5%), arterial thromboembolic events (ATEs) (2.5%), hypertension (27.1%), congestive heart failure (CHF) (1.2%), wound healing complications (3.2%), GI perforations (1.9%), posterior reversible encephalopathy syndrome (PRES) (0.2%), Neutropenia (43.1%), venous thromboembolic events (VTEs) (6.7%), fistulae (Non-GI) (1.0%), thrombotic microangiopathy (<0.1%), pulmonary hypertension (0.1%), hypersensitivity and infusion reactions (27.6%), gallbladder perforation (<0.1%), peripheral sensory neuropathy (25.4%), non-CHF/ATE cardiac disorders (2.7%), osteonecrosis of the jaw (ONJ) (0.1%), and thrombocytopenia (26.8%). 11 This study reported bleeding/hemorrhage (2.9%), hypertension (4.9%), neutropenia (8.9%), vaginal fistula (0.5%), chills (0.5%), and thrombocytopenia (3.5%) in line with earlier studies but at a lower frequency. The most frequently reported AEs in this study includes diarrhea (11.3%), asthenia (10.3%), headache (8.9%), anemia/decreased hemoglobin (8.4%), vomiting (7.9%), pain (7.4%), pyrexia (5.4%), fatigue (3.9%), dry mouth (3.0%), insomnia (2.5%), alopecia (2.5%), decrease appetite (2.5%), nasopharyngitis (2.0%), and eye pain (2.0%). This study reported ADAs in 2 (1.75%) patients which is slightly higher than the US-approved Bevacizumab (0.63%) and less than the EU-approved Bevacizumab (2.5% 12 and 4.2% 13 ). Like other immunogenic proteins, the incidence of immunogenicity is most likely to be affected by the use of concomitant therapy, such as myelosuppressive chemotherapeutic agents and extent of prior therapy. The results of our phase IV, post-marketing study confirm the manageable safety profile, efficacy, and immunogenicity of Hetero-Bevacizumab, support the use of Bevacizumab in both the first- and second-line treatment of solid tumors in the approved indications by the FDA, and establish the relevance of findings of our randomized phase III studies. 14 In our study, the overall response rate was 40.9%, which is comparable to our phase III study (ORR was 46.9%) and to previously published clinical studies which reported ORR as 30% to 40%, depending on the prescribed regimen.15-21
The development of biosimilars offers an affordable, safe, and effective alternative to potent biological therapies and comparative clinical studies help generate evidence in that direction. The results of this study provide post-marketing clinical evidence for the biosimilar Hetero-Bevacizumab in relation to the efficacy, safety, and immunogenicity data in patients with solid tumors, suggesting a possibility for interchangeability of usage.
Conclusion
Based on the study results, it can be concluded that Herero’s Bevacizumab (Cizumab) was safe, well-tolerated, and efficacious. In conjunction with the data obtained in the phase III trials conducted on Hetero-Bevacizumab, this product can be used interchangeably with approved and available Bevacizumab brands for the management of patients with approved indications in solid tumors. This data provides a trend of its safety profile and efficacy of Bevacizumab in real-world scenarios of prescribed settings and is consistent with our phase III study and published literature.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Concept and design of trial, regulatory approvals: S.S., P.T., S.C., B.R.; Project management: S.K. and P.T.; Manuscript drafting and revision, statistical analysis, data interpretation and conclusion analysis: S.S., S.C., L.T. and R.V. All the investigators in Bevacizumab investigator group were responsible for conducting trial as per study protocol, site SOPs and relevant applicable regulations. All authors contributed to the study and approved the final manuscript.
Availability of Data and Materials
Data supporting the findings are presented within the manuscript and additional datasets used are available from the corresponding author on request.