
Abstract
Identifying individual mechanisms involved in complex diseases, such as cancer, is essential for precision medicine. Their characterization is particularly challenging due to the unknown relationships of high-dimensional omics data and their inter-patient heterogeneity. We propose to model individual gene expression as a combination of unobserved molecular mechanisms (molecular components) that may differ between the individuals. Considering a baseline molecular profile common to all individuals, these molecular components may represent molecular pathways differing from the population background. We defined an infinite sparse graphical independent component analysis (isgICA) to identify these molecular components. This model relies on double sparseness: the source matrix sparseness defines the subset of genes involved in each molecular component, whereas the weight matrix sparseness identifies the subset of molecular components associated with each patient. As the number of molecular components is unknown but likely high, we simultaneously inferred it and the weight matrix sparseness using the beta-Bernoulli process (BBP). We simulated data from a double sparse ICA with 10/30 components with specific sparseness structures for 100/500 individuals and 500/1000/5000 genes with different noise variance levels to evaluate the reconstruction of the latent structures by our model. For all simulations, the isgICA was able to reconstruct with higher accuracy than 2 state-of-the-art methods (ica and fastICA) the number of components, the weight and source matrix sparsenesses (correlation simulated/estimated >.8). Applying our model to the expression of 1063 genes of 614 breast cancer patients, the isgICA identified 22 components. According to the source matrix, 7 of these 22 components seemed to be specifically related to 3 known molecular pathways with a prognostic effect in early breast cancer (immune system, proliferation, and stroma invasion). This proposed algorithm provides an insight into individual molecular heterogeneity to better understand complex disease mechanisms.
Keywords
Introduction
A comprehensive understanding of the molecular mechanisms of complex diseases, such as cancer, is one of the main current challenges to develop precision medicine. Identifying these mechanisms from omics data, such as gene expression, is challenging due to the complex relationships in various molecular pathways involving hundreds or thousands of actors (eg, genes) and the relatively small number of patients in the available data sets.
To limit the curse of dimensionality, the identification of non-observed high dimensional omics data structures, which provide an insight into the molecular mechanisms, is often performed using latent variable models 1 (LVM) for blind source separation/deconvolution, including principal component analysis (PCA), independent component analysis (ICA), or factor analysis (FA). To identify independent molecular components, we based our work on the ICA model, and we use below the corresponding terminology, that is, the source and the weight matrices corresponding to the parameters representing the association of the components with the genes and the observations, respectively.
To interpret the identified structures as molecular mechanisms or pathways, sparse methods may be used to select a subset of the omics variables associated with each component, similarly to the graphical factor model proposed by Yoshida and West. 2 As illustrated by Figure 1, the sparseness structure of the source matrix may be considered as a hypergraph matrix. The hypergraph approach is a generalization of the graph methods considering higher-order interactions of the nodes (eg, genes) to model complex relationships, 3 which are represented by different (potentially overlapped) subsets of nodes associated to different hyperedges. Each of these latent structures may represent different molecular mechanisms such as pathways, associated to only a subset of the gene expressions. Several sparse approaches have been proposed, especially in the Bayesian framework, imposing sparseness on the source matrix using the spike and slab prior,4,5 Indian Buffet Process, 6 Laplace prior, 7 or the horseshoe prior. 8

Independent component analysis matrix sparseness structure interpretation as a hypergraph. The source matrix sparseness (top) represents K molecular components (hyperedges) associated with different gene (node) combinations that may represent different molecular mechanisms or their alterations. The weight matrix sparseness (bottom) defines different profiles defined by individual combinations of these molecular components to characterize patient disease heterogeneity.
Although this approach is suitable to give an insight into mechanisms common to all individuals, it is known in oncology that the tumor may result from different molecular mechanisms (or different alterations/state of the same mechanism) across different patients that complicate the development of drugs applicable to broad cancer populations. A natural way to consider this inter-individual heterogeneity with LVM is to impose a second sparseness structure to the weight matrix, associating the individuals to the components. Again, the corresponding sparseness structure may be considered as a hypergraph (Figure 1), and in which this time, each hyperedge represents the subset of individuals presenting the corresponding molecular mechanisms (see Figure 1).
As the number of molecular mechanisms is unknown (but likely large), and the number of their possible alterations may grow rapidly with the number of individuals, one would prefer not to fix the dimension of the model but to infer the number of components present in the studied population. These 2 modeling perspectives may be considered simultaneously using the beta-Bernoulli process (BBP) 9 as a prior on the hypergraph matrix of the weight matrix.6,10 The rationale behind this approach is to consider a prior on the infinite-dimensional model space assigning only a finite number of 1 in the hypergraph matrix almost surely (therefore a finite number of sparse components) in a finite sample. 11 In other words, this approach considers that there is an infinite number of molecular alterations, but only a subset is present in our finite sample. This approach has the appealing property to allow the model’s complexity to grow with the number of observations under the regularization of the BBP hyperparameters.10 -12
While previously mentioned works focused on sparse coding of the weight matrix or of the source matrix, we propose to impose sparseness on the mixture weight matrix and on the source matrix. To our knowledge, this is the first study imposing this double sparseness in an infinite-dimensional model and proposing an optimization procedure to improve the reconstruction of the underlying latent structures. However, the interpretation of the resulting components remains complex and hazardous because they may represent different molecular mechanisms/pathways or their different alterations among the patients. Instead of precisely characterizing these molecular mechanisms, we propose to identify “alterations” from a baseline molecular profile. Our choice was motivated by the assumption that the differential disease progression or drug resistance results in a mixture of multiple molecular alterations, which could be different between the patients. We imposed this baseline constraint by enforcing the first component of the weight matrix to be a vector of ones, that is, all individuals are associated with this component, representing the molecular background of the population (that we named the baseline molecular profile). We also consider a noise component (such as the noisy ICA 13 ) to capture the specific individual background (shared with no other individuals) and measurement error.
In this work, we first assessed the ability of our isgICA to reconstruct the weight and the source sparseness structures through a simulation study. We compared our approach for the identification of the number of components and for the reconstruction of the matrices to state-of-the-art algorithms. Finally, we applied our method to model the gene expression heterogeneity in a large gene expression dataset of tumors from breast cancer patients included in clinical trials of anthracycline-based chemotherapy and illustrate the relevance of this algorithm to blindly identify relevant known breast cancer gene expression signatures.
Methods
Infinite sparse graphical independent component analysis (isgICA)
Let

where
We used this approach to impose sparseness on

As the number of unobserved molecular mechanisms is unknown but likely high, we consider a nonparametric ICA with an infinite number of components (ie, K = ∞). The beta-Bernoulli process (BBP) is a suitable nonparametric prior for binary matrix (
Baseline profile and isgICA model
We define a baseline profile as a non-sparse latent component, that is, a component associated with all individuals; that is,
Complete model formulation
We considered conjugate priors for the elements of the model matrices, which allow for posterior analytical calculation and straightforward inference. The graphical representation of the proposed model is illustrated in Figure 2.
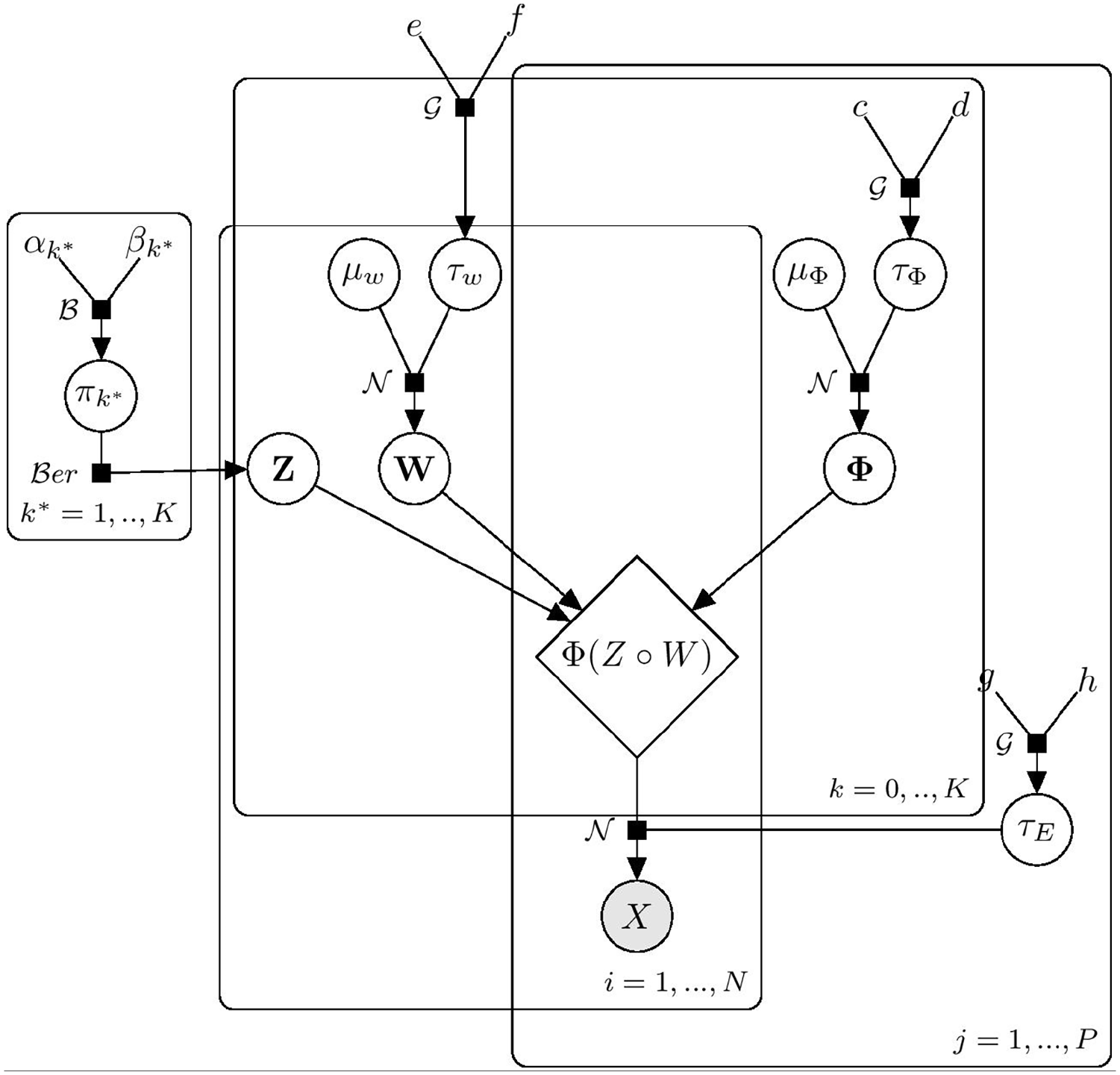
Graphical model representation of the infinite sparse independent component analysis. Observed variables are denoted with shaded nodes, while unobserved variables are shown as white nodes.
The complete model is expressed as:

where
Consistent estimation of the noise precisions (
Parameter inference and hyperparameter tuning
As the posterior computation using MCMC is notoriously slow when the number of parameters is high, we used variational Bayesian (VB) inference under the mean-field assumption to approximate the true posterior distribution.15,16 We derived the variational evidence lower bound of the likelihood (ELBO) and the variational parameter update equations in Appendix A1.
For a computational purpose, we used the truncated beta process for the inference, with a maximum number of components noted
Due to the lack of a simple analytical form of the conjugacy between the prior of the beta distribution and the beta distribution for its moments, we tuned the
Standard whitening ICA
We evaluated the ability of our method to reconstruct the matrix sparseness structures from simulated data sets in comparison with state-of-the-art algorithms. Due to computational issues for our larger scenario (time and/or memory), we used the standard whitening ICA model implemented in the ica 20 and the fastICA 21 R packages, and pre-selected the components with eigenvalue higher than one. The eigenvalue criteria specifies that only components with eigenvalues larger than 1 should be preserved since each component should explain the variance of a single variable.
Results
The simulations, the parameter optimization, and the data visualization were performed using R software (version 3.6.0). The R codes and data are available on https://github.com/Oncostat/isgICA .
Synthetic data
Scenarios
We simulated synthetic datasets from equation (3), according to different scenarios (see Table 1) with
Simulation scenarios. N, P, and

For all scenarios, the elements of the source matrix Φ were generated for each component from Gaussian distributions with variance equal to one, and different means (from −3 to 3) to evaluate if the model may identify components with specific patterns, such as mainly positive or negative values, or both (for means closed to 0). Random blocks were generated to assign sparseness structure to this matrix presented by the Figure 5). The elements of the weight matrix W were drawn from a standard Gaussian distribution (mean = 0, variance = 1). We simulated 10 data sets for each scenario.
Performance criteria
As the standard ICA, our model is identifiable up to scaling, sign reversion, and column permutation.
22
To evaluate the model reconstruction, we aligned the estimated components to the simulated ones using as a distance the mean of the absolute Pearson correlation coefficients of each pair of the column of the simulated source matrix
The mean of the absolute Pearson correlation coefficients between the simulated Φ and
According to the component ordering defined previously, the reconstruction of the sparseness structure of the weight matrix (except the baseline profile, ie, Z*) was assessed with the accuracy criterion, defined by:
Latent structure reconstruction
We first evaluated the ability of the algorithms to identify the number of components (Figure 3 and Table 2). The isgICA recovered the exact number of latent components in the majority of the simulations, but it underestimated the number in the lower dimension scenarios (P = 500), especially when the number of observations was low with respect to the number of component (N = 100, K = 30). This behavior was slightly more apparent when the noise variance was increased. The number of components selected with the classical whitening ICA using the eigenvalue method increased quickly with the dimension (P/N ratio) to the maximal number of components allowed by this approach (minimum between N-1 and P-1). It also slightly increased when the noise variance was raised in our algorithm.
Number of identified components (median [2.5%-97.5%] percentiles) using the eigenvalue method for the standard ICA and by the isgICA. N, P, and

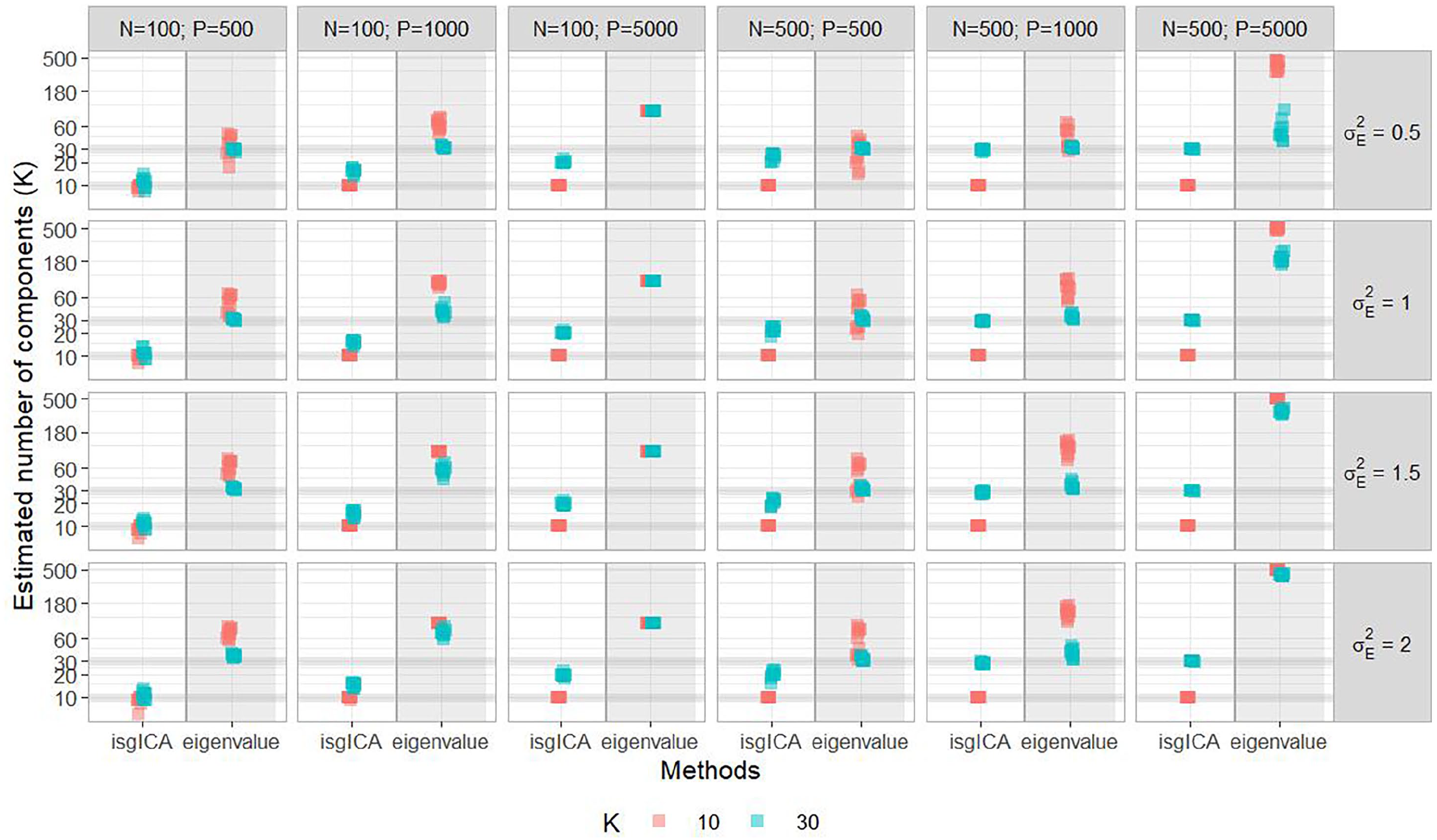
Reconstruction of the number of latent components between isgICA and the standard ICA using the eigenvalue criteria with 10 (red) or 30 (blue) simulated components (K). Each row corresponds to the scenarios with different noise variances (
Due to this over-decomposition using the eigenvalue method in the majority of the scenarios, the calculation of the reconstruction criteria for the ica and fastICA was not possible. In this case, we selected the 10 estimated components (or 30, according to the scenario) for which the source components were the most correlated to the simulated ones. We also performed an oracle sensitivity analysis, fixing a priori the number of components to the simulated ones for these 2 approaches, in order to have a comparison of their reconstruction to the blind reconstruction of the isgICA (see results in Appendix A2, Figures A1 and A2)
The isgICA outperformed the ica for the reconstruction of the source matrix (Φ) when the number of components was estimated using the eigenvalue method (Figure A1). This difference was less clear for the fastICA (correlation to simulated source matrix equal to .818 [.191, .934] (median [2.5%-97.5%] percentiles) for isgICA, .767 [.513,.982] for fastICA, and .189 [.0.28,.834] for ica), especially due to the lower performance of the isgICA in the scenarios with the lowest dimensions (N = 100/P = 500, and N = 500/P = 500). The performance of all methods decreased with the increase of the noise variance, but the isgICA was less impacted for high dimensional scenarios. The oracle fastICA and ica models presented in the majority of the scenarios a performance similar to the isgICA, excepting in the scenarios where the isgICA underperformed, that is, in the case of low dimension.
For all the scenarios, the isgICA outperformed the other methods for the reconstruction of the weight sparse matrices
The isgICA results are summarized in the Figure 4. The ability of the isgICA to reconstruct the sparseness structure of the weight matrix (

Reconstruction of the weight sparseness structure (Z), of the weight sparse matrix (Z°W) and sources matrix (Ф) according to different noise variances in rows with 10 (red) and 30 (blue) simulated components (K) and different dimensions (N individuals and P genes) in columns: the accuracy of the reconstruction of Z (first criteria), the mean absolute correlation of
To illustrate the reconstruction ability of the isgICA, the Figure 5 shows the reconstruction of the sparse weight matrix (

Visual representation of the reconstruction of the weight sparseness structure (Z, top row, accuracy of .982) sparse weight structure (W°Z, middle row, correlation of .999) and sources(Ф, bottom row, correlation of .873) from a diagonal structure and with N = 500, P = 5000, K = 10 and
It can be noted that the ica method did not converge for 2.9% (14/480) simulations of the high dimension scenario (N = 500, P = 5000, K = 10) due to the large number of components identified using the eigenvalue method. Therefore, the result of this method is slightly overoptimistic and should be interpreted accordingly (including for the context of a real application).
The gain of precision for the (blind) matrix reconstruction and convergence rate for the isgICA relatively to the other methods came at the cost of a larger computational time relatively to the eigenvalue method (few minutes to several hours), that increases quickly with the dimension (Appendix A2, Figure A3).
Application: Early breast cancer data
We applied our method to publicly available gene expression data obtained from tumor biopsies in 614 breast cancer patients that were included in clinical trials of anthracycline-based chemotherapy,24,25 available in the biospear R package. 26 The expression data of 22 277 probes (Affymetrix array) was preprocessed via frozen robust multiarray 27 and cross-platform normalization. 28 Probes were filtered if the interquartile range ⩽1. The remaining 1689 probes were standardized and then filtered with the package jetset 29 to retain a single probe by gene, resulting in a final dataset including the expression of 1063 genes. As in Belhechmi et al, 30 we mapped to probes to 3 molecular signatures with a prognostic effect in early breast cancer (Immune System, Proliferation, and Stroma invasion 31 ) and one without (SRC activation signature 31 ), all the other probes were categorized as “Others”.
Figure 6 presents the hypergraph matrix of the individual heterogeneity. The model identified 22 non-zero components (including the baseline profile).

Hypergraph matrix of the individual heterogeneity (weight sparseness structure) extracted from the breast cancer dataset using the infinite sparse graphical independent component analysis with baseline profile. The model identified 22 components (including the baseline profile).
To investigate the molecular relevance of these results, we overcame the problems of sign and scale reversion by ranking the absolute source element values amongst each component to identify the most contributive genes to each identified molecular alterations. Figure 7 shows the distribution of the absolute values of the source matrix elements of each component according to the different breast cancer signatures. The proliferation-based signature, immune-system signature, and stroma-related signature seemed to be related to the components 2/5, 4/7/8/15, and 3, respectively. The SRC, which was picked as a “negative control” signature in Belhechmi et al, 30 was not straightforward to map to a particular molecular component.

Distribution of the absolute value of the source matrix elements associated with the probes of genes associated with 3 molecular signatures with a prognostic effect in early breast cancer (GGI, Immune2, and Stroma2) and one without (SRC activation signature). All the other genes were categorized as “Others”. A proliferation-related signature, immune-related signature, and stroma-related signature seemed to be related to components 2/5, 4/7/8/15, and 3, respectively. The SRC was not related to one specific component.
Discussion
In this paper, we proposed a novel approach to characterize inter-patient heterogeneous molecular mechanisms. To our knowledge, this is the first approach that assumes that the molecular profile of each patient is a mixture of different molecular components, which can be shared with the other patients. We modeled these components as alterations from a baseline molecular component shared by all individuals, representing the mechanisms common to all patients, while the noise captures the individual molecular background. Assuming that each molecular component represents alterations of a molecular pathway or a group of related pathways, this approach may help us to understand molecular mechanisms and identify potential targets for drug development.
We illustrated the concept using a gene expression dataset of breast cancer tumor samples from patients included in clinical trials of anthracycline-based chemotherapy. The correspondence of the identified molecular profiles with known molecular pathways that play a prognostic role in early breast cancer (proliferation, immune system, and stroma pathway) suggests that our approach may help characterize the molecular context of particular subpopulations.
In the simulation study, our method was capable of blindly identifying the true number of components and their (sparseness) structures, up to scaling and sign reversion, which are well-known identifiability issues in standard ICA. To alleviate these issues, we proposed to use the absolute values of the source elements to identify the most contributive genes to each component for molecular interpretation. Comparing to 2 other popular ICA algorithms (fastICA and ica), our model better reconstructed blindly the number of components and the weight and source matrices, and had a similar performance when we a priori fixed the true number of components in these 2 algorithms.
Our algorithm was able to provide a better reconstruction performance of the weight sparse matrix
As illustrated by our benchmark, the performance of our algorithm came at the cost of an important computational time that could be a practical limitation. A first next step will be to re-implement the current algorithm with GPU computation to scale-up to large datasets.
We showed that this approach is able to identify blindly components deviating from a baseline profile. Future research will focus on improvements for their identifiability and interpretability, including the integration of additional external information. We expect that some mixture weight components can reconstruct observed individual variables not considered by the model. It could be possible to extend this model, fixing the elements of some weight components to the values of observed individual variables, which may be relevant to explain the gene expressions (eg, gender, age, tumor stage). Beyond the adjustment for known characteristics, this extension could be used to perform differential analysis adjusted for unobserved individual characteristics. Moreover, while the bulk sequencing data results of a mixture of several elements (not only tumor cells, but also healthy tissue 33 or tumor microenvironment cells 34 ), other sources of data such as reference molecular profiles could be used to improve the identifiability and interpretability of the components. Another interesting way to integrate external information is to consider the patient characteristics as explanatory variable of the sparseness structure of the source matrix to model different states of the graph, as suggested by Wang et al. 35
The joint modeling of our isgICA and a clinical outcome, such as patient survival, could be of particular interest for precision medicine, favoring the identification of independent molecular profiles more specific to the patient prognostic. This extension will support the estimation of component/treatment interaction in the survival model to highlight pathways related to treatment response for the precision medicine context.
Finally, as proposed in the wide literature of omics data deconvolution methods, our approach may also be extended to other non-Gaussian omics data, such as count (raw RNAseq, proteomics) or binary (mutations) data using different link functions.
Conclusion
We developed an isgICA model with a baseline profile to characterize blindly the individual heterogeneity from this baseline profile in a high-dimensional setting. This approach illustrates a novel concept for the identification of composite molecular profiles which could be key to understanding the different mechanisms of disease and identify potential targets to develop new treatments.
Footnotes
Appendix
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SLR developed the model, and takes responsibility for the R programs and the accuracy of the data analysis. SLR drafted the manuscript; DD and SM contributed to a critical revision of the manuscript for important intellectual content, supervised the study equally, and gave final approval. All of the authors read and approved the final manuscript.