
Abstract
Long non-coding RNAs (lncRNAs) are a large and diverse class of transcribed RNAs, which have been shown to play a significant role in developing cancer. In this study, we apply integrative modeling framework to integrate the DNA copy number variation (CNV), lncRNA expression, and downstream target protein expression to predict patient survival in breast cancer. We develop a 3-stage model combining a mechanical model (lncRNA regressed on CNV and target proteins regressed on lncRNA) and a clinical model (survival regressed on estimated effects from the mechanical models). Using lncRNAs (such as HOTAIR and MALAT1) along with their CNV, target protein expressions, and survival outcomes from The Cancer Genome Atlas (TCGA) database, we show that predicted mean square error and integrated Brier score (IBS) are both lower for the proposed 3-step integrated model than that of 2-step model. Therefore, the integrative model has better predictive ability than the 2-step model not considering target protein information.
Introduction
Several evidences highlight the emerging impact of long noncoding RNAs (lncRNAs) in cancer progression.1-4 The aim of this study is to identify the predictive capability of some oncogenic lncRNAs in tumor progression and prognosis of breast cancer.
Breast cancer is the most common malignancy and the leading cause of cancer death in women. By focusing on a single type of genetic alteration such as copy number variation (CNV), scientists have identified significant genes that may contribute to cancer progression.5-8 Due to its complexity, the study of cancer should focus on incorporating data from multiple platforms ranging from genes, transcripts, and proteins found in cancer cells, 9 to whole biological systems, represented by molecular pathways and cell populations. 10 The integration, where multiple levels of omics data (ie, CNV, methylation, and gene expression) are gathered from the same subjects and analyzed, is known as vertical integration.10-12
In this study, we introduce an easy and simplified way to integrate multiple omics data to show that the survival prediction due to the presence of lncRNAs increases significantly in breast cancer. We consider the genomic platform such as CNV, mRNA expression, proteomic platform such as protein expression, and the phenotype such as the survival of the patients. This study focuses only on the lncRNA expressions from The Cancer Genome Atlas (TCGA) breast cancer data. We consider the target protein expressions as proteomics data.
An Integrative Model
We consider a 3-stage model here. Suppose that n is the number of patients, p is the number of lncRNAs, and L is the number of CNV expressions.
The mechanistic model for each lncRNA can be expressed as

where
Next, the downstream target protein of each specific lncRNA was identified from PubMed articles, TCGA RNA-Seq database, and other extensive analyses such as differential expression analysis. The mechanistic model for each protein (for every lncRNA) can be expressed as

where
The clinical component part models the effect of the mechanistic parts of the genes on a clinical outcome of interest and can be written as
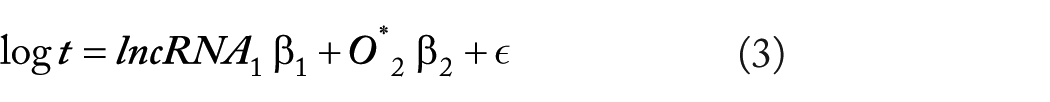
where
The variable
Assumptions such as
In the presence of right censoring, we observe the tuple
To quantify the prediction accuracy, we consider a standard comparative predictive approach Brier score (BS) 13 which uses the predicted survival times

where
Nevertheless, we also compute the prediction square error by comparing the observed data and their posterior predicted values.
From TCGA database, we consider the information of 222 breast tumor samples with their survival data. We observe that at least 82% data are right censored.
Along with the clinical observations, we also collected measurements of 12 lncRNA expressions (Table 1). Among those, we found the CNV information available for 9 genes (or lncRNAs). We also consider 64 target protein expressions for these genes.
The lncRNA considered for our experiment.
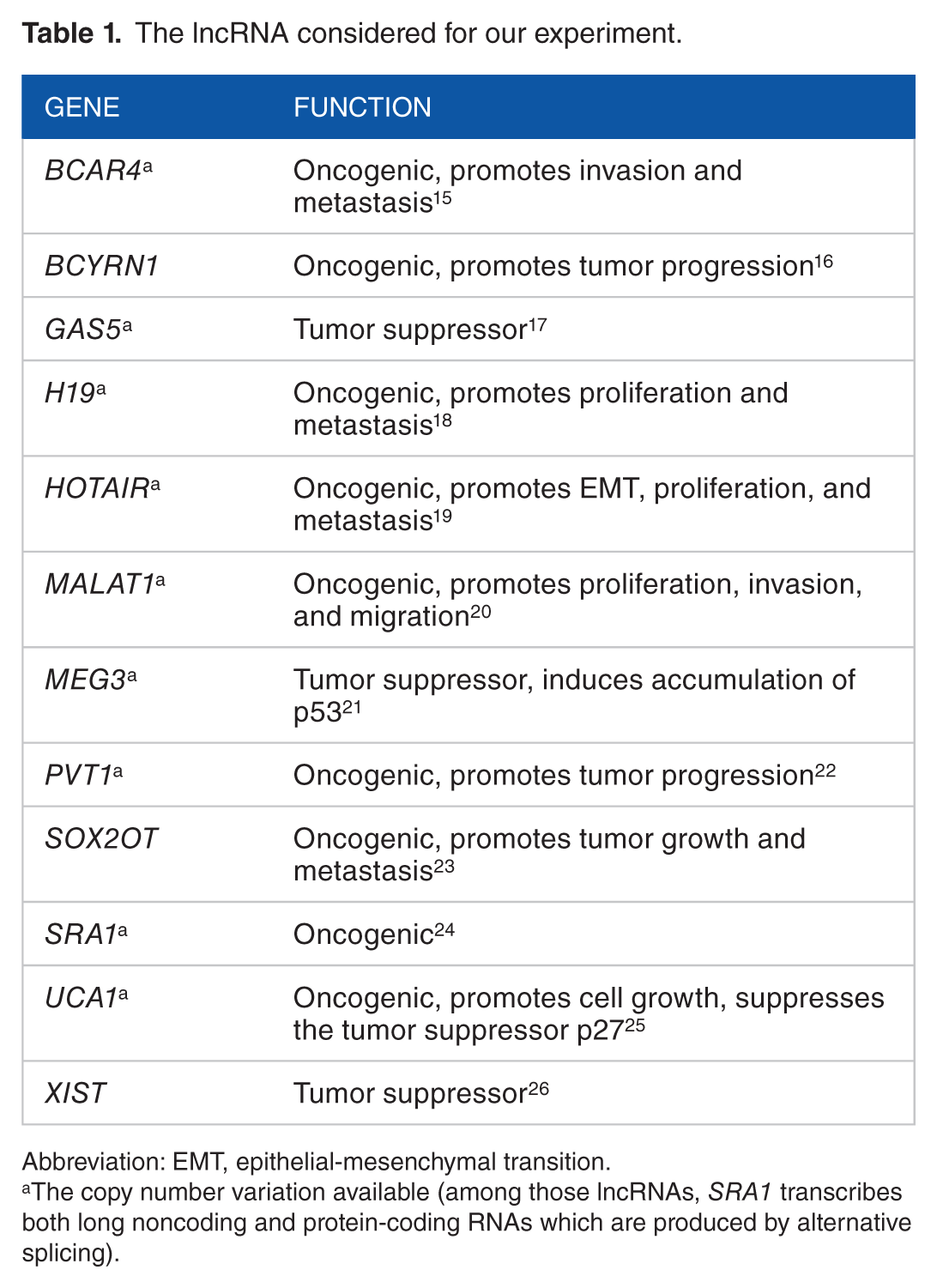
Abbreviation: EMT, epithelial-mesenchymal transition.
The copy number variation available (among those lncRNAs, SRA1 transcribes both long noncoding and protein-coding RNAs which are produced by alternative splicing).
We apply the integrative modeling in these data and obtain the results shown in Table 2. We notice that the mean squared prediction error and IBS are both lower for the proposed model than for the 2-stage model after omitting the protein expressions from the analysis.
MSPE and IBS for fitted models in TCGA breast cancer data.

Abbreviations: IBS, integrated Brier score; MSPE, mean squared prediction error; TCGA, The Cancer Genome Atlas.
In this article, we have shown that when the contribution of lncRNA’s target protein expression measurement is not ignored, then the survival prediction has improved dramatically. Toward this, we have developed a simple yet integrative modeling strategy which borrows strengths from all 3 platforms such as DNA CNV, mRNA expressions for the long noncoding genes, and their target protein expressions to predict the survival of the subjects. We have shown that this integrated model outperforms its closest competitor.
Footnotes
Acknowledgements
The authors thank the editor and the reviewers for their helpful suggestions which substantially improved this paper.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: T.R.S. was supported through the NIH T32 Training grant (PI: Dr Raymond J Carroll); A.K.M. and B.K.M. were supported through NIH R01CA194391 (PI: Dr B.K.M.).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
TRS, AKM, and BKM designed the study. AKM and YN collected and analyzed the data. TRS and AKM wrote the manuscript.