
Abstract
Background:
Pseudogenes are sequences that have lost the ability to transcribe RNA molecules or encode truncated but possibly functional proteins. While they were once considered to be meaningless remnants of evolution, recent researches have shown that pseudogenes play important roles in various biological processes. However, the studies of pseudogenes in the silkworm, an important model organism, are limited and have focused on single or only a few specific genes.
Objective:
To fill these gaps, we present a systematic genome-wide studies of pseudogenes in the silkworm.
Methods:
We identified the pseudogenes in the silkworm using the silkworm genome assemblies, transcriptome, protein sequences from silkworm and its related species. Then we used transcriptome datasets from 832 RNA-seq analyses to construct spatio-temporal expression profiles for these pseudogenes. Additionally, we identified tissue-specifically expressed and differentially expressed pseudogenes to further understand their characteristics. Finally, the functional roles of pseudogenes as lncRNAs were systematically analyzed.
Results:
We identified a total of 4410 pseudogenes, which were grouped into 4 groups, including duplications (DUPs), unitary pseudogenes (Unitary), processed pseudogenes (retropseudogenes, RETs), and fragments (FRAGs). The most of pseudogenes in the domestic silkworm were generated before the divergence of wild and domestic silkworm, however, the domestication may also involve in the accumulation of pseudogenes. These pseudogenes were clearly divided into 2 cluster, a highly expressed and a lowly expressed, and the posterior silk gland was the tissue with the most tissue-specific pseudogenes (199), implying these pseudogenes may be involved in the development and function of silkgland. We identified 3299 lncRNAs in these pseudogenes, and the target genes of these lncRNAs in silkworm pseudogenes were enriched in the egg formation and olfactory function.
Conclusions:
This study replenishes the genome annotations for silkworm, provide valuable insights into the biological roles of pseudogenes. It will also contribute to our understanding of the complex gene regulatory networks in the silkworm and will potentially have implications for other organisms as well.
Introduction
Pseudogenes are sequences that are similar to those of functional genes but have lost the ability to encode normal proteins or RNA transcripts.1 -3 There are 2 main ways to produce pseudogenes: duplicated pseudogenes (DUPs) and processed pseudogenes (also known as retropseudogenes (RETs)). 4 DUPs are produced by the loss of their original gene function due to the generation and accumulation of sequence mutations during replication, and RETs are produced by reverse transcription of processed mRNA, followed by integration into the genome. 5 Due to the different mechanisms for the formation of these 2 major types of pseudogenes, the main difference in their sequences concerns whether the non-coding regions in the gene such as introns are retained.
With the improvement in research methods, especially the development of bioinformatics and high-throughput sequencing technology, researchers have begun to realize that pseudogenes may have important biological significance.2,6 For example, recent studies have shown that some pseudogenes can encode proteins that are shorter than the parental functional genes but do have functions. 7 Some pseudogenes regulate gene expression through transcription into RNA-mediated regulatory species such as antisense RNA, siRNAs (Small interfering RNA), miRNA (MicroRNA), and snoRNA (Small nucleolar RNA). 8 Some pseudogenes record the evolution of genome sequences at the molecular level, thereby providing ideal materials for genome evolution research. 9 Because pseudogenes are widely present in various organisms and play diverse roles, it is significant to study pseudogenes for understanding organic evolution, genome evolution, and genome regulation.
Since Jacq et al 10 first introduced the term “pseudogenes” in 1977 to describe the truncated ribosomal genes found in the study of Xenopus laevis, researchers have gradually begun to study the functions of pseudogenes. 11 To date, the study of pseudogenes has made important progress in common model organisms such as humans, mice, and fruit flies.12 -14 Silkworms have been domesticated for >5000 years, and they are the only insects that have been domesticated by humans. 15 The pupae are used for silk production in traditional agriculture and more recently as a protein additive raw material for livestock. 16 In addition, silkworms can also be used for commercial production of important biomedical and industrial biomaterials through genetic engineering.17,18 Silkworms are often employed in biological research due their low maintenance cost, fewer ethical constraints, lack of biological hazard risk, and similarity to humans in terms of sensitivity to pathogens and the impact of drugs. 19 Silkworms have always been considered an excellent model organism for studying physiology, biochemistry, developmental biology, neurobiology, and pathology.20,21
The silkworm pangenome project have provided a large-scale genetic resource (such as genome assemblies, genomic variations and traits) for research communities, therefore, laid a foundation to study the relationships between traits and genomes. 15 However, most genomic variations are located outside of protein-coding genic regions, therefore, it’s urgent to obtain more comprehensive genome annotations, such as miRNA,22,23 lncRNA, 24 smORF, 25 and pseudogenes. Researches involves in pseudogenes silkworms are limited. Kondo et al 26 found that 5 of 38 bombyxin are pseudogene candidates. Vega et al 27 proposed that the U1 family, which related to mRNA precursor processing in silkworm, may be derived from an ancestral pseudogene. Fotaki and Iatrou 28 also identified the transcriptional activity and biological function of a chorion locus pseudogene in silkworm. However, these studies were based on single or a few pseudogenes, and there were no genome-wide systematic studies yet.
Here, we employed the reference genome assembles of the silkworm and their close relatives, high-confidence protein sequences, and 832 transcriptome datasets with detailed and reliable information to identify the pseudogenes of the silkworm at the genome-wide level. We then explored the temporal and spatial expression patterns of these pseudogenes through transcriptome analysis. This study provides a theoretical basis for the further analysis of pseudogenes in the silkworm as well as a reference for other species for which pseudogene research at the whole gene level has not yet been carried out.
Methods
Genomic and transcriptomic dataset collection
Genome and gene annotation datasets for Bombyx mori were downloaded from SilkBase (http://silkbase.ab.a.u-tokyo.ac.jp/cgi-bin/download.cgi). 29 In addition to the silkworm, this study also used datasets from species that are closely related to the silkworm, including the model organism Drosophila melanogaster (GCF_000001215.4) as well as B. mandarina (GCF_003987935.1), Anduca sexta (GCF_014839805.1), and Dendrolimus kikuchii (GCA_019925095.2). The above genome and annotation datasets were downloaded from the National Center for Biotechnology Information (NCBI).
To explore the spatio-temporal expression profiles of silkworm pseudogenes, we collected the RNA-Seq datasets from >1200 silkworm samples deposited in the NCBI SRA database. After manual inspection of these records from the NCBI database or the corresponding articles, we obtained 832 samples with unambiguous tissue or developmental stage information (Supplemental Table S1).
Genome wide identification of pseudogenes in the silkworm genome
First, we generated a high-confidence reference protein sequence dataset for subsequent analyses with the following steps: (1) gene models from the silkworm and closely related species were filtered for those with incomplete coding sequences (CDS); (2) proteins from SwissProt were incorporated after removing fragmental or transposon-related proteins; (3) redundant proteins were removed by cd-hit.
The identification of pseudogenes was divided into the following steps: (1) repeatmasker (http://www.repeatmasker.org) was used to mask the repetitive sequences in the silkworm genome sequence, and then CDSs of the high-confidence genes of the silkworm were removed. (2) the blastx function of diamond 30 (version: v0.9.24.125) was used to align the high-confidence reference protein sequence dataset obtained above to the silkworm genome sequence after masking the repetitive sequences and high-quality CDS. (3) The genomic regions aligned to a reference protein as well as their upstream and downstream 10 kbp regions were extracted as candidate pseudogene regions for subsequent accurate genewise alignment. Since the reference protein sequences contained the sequences of related species and the SwissProt database, we use the blast score value to determine the best hit result of each region. (4) According to the obtained best hits, the Genewise 31 (version: wise2.4.1) was used to compare each of the above proteins with the candidate regions on the genome to which they were aligned, and then the genewise results were parsed to obtain the pseudogene loci.
Classification and distributions of pseudogenes
Following the previous studies, 32 we divided the pseudogenes into 4 categories: duplications (DUPs), unitary pseudogenes (Unitary), processed pseudogenes (retropseudogenes, RETs), and fragments (FRAGs). Among these, Unitary means a pseudogene that was originally functional with a single copy gene in the genome that has undergone spontaneous mutation in the coding region or regulatory region, resulting in the gene being unable to be transcribed or translated. FRAG means that its corresponding parent gene has multiple exons, but it can only be aligned to one of the them, that is, the pseudogene originated from a single exon coding region. DUP, Unitary, and RET can also be subdivided into full and truncated types according to whether the coverage is lower than 20% in the alignment results of the pseudogenes to their corresponding parent genes.
Pseudogenic background is referred as the genomic environment of the pseudogene, that is, whether the pseudogene is located in the exon and/or intron of protein-coding genes or intergenic regions. To compare the locations of pseudogenes and protein-coding genes, the trmap (v0.12.6) analysis was performed, which uses a similar scheme to the GffCompare 33 (0.12.6) transcript classification priority. The pseudogene density (the ratio of total pseudogenes to total genes) in each 100 kb intervals was calculated and plotted.
Analyzes of pseudogenes and their parent genes
The distribution of identities of pseudogenes and their parent genes, and since the parent gene of Unitary is missing in the silkworm, this distribution did not include the unitary pseudogenes. We obtained the functional annotation results of pseudogenes based on the parent genes. GO and KEGG annotations of parent genes were performed by Interproscan 34 (version: v5.59_91.0) and KoFamScan 35 (version: v1.3.0). Enrichment analyses of 3 types of pseudogenes (DUPs, RETs, and Unitary) were performed using their parent genes as proxy.
Evolutionary analyses of pseudogenes
Considering that the sites after the pseudogene causing mutation site (premature stop codon or frameshift site in the pseudogene) were no longer subject to selection pressures, the divergence times of pseudogenes and their parent genes could be calculated using these segments. The pseudogenes without early termination codons or frameshift sites were excluded in this analysis. We obtained the sequences behind the premature stop codon or frameshift site in the pseudogene according to alignments of the pseudogenes and their parent sequences. We then filtered the results as follows: the length of the sequence is less than 50 bp, or the proportion of the alignment length is <50%. Distmat (http://www.bioinformatics.nl/cgi-bin/emboss/distmat) was used to calculate the distance
We also analyzed the selection pressure of the pseudogenes: (1) the CDSs of each pseudogene and their corresponding parent gene (only the silkworm genes) were aligned by muscle 36 (version: v5.1), and then the Ka/Ks value of each pseudogene and its corresponding parent gene was calculated by the YN00 module of PAML package 37 (version: v4.10.6). Finally, the distributions of selection pressures on the whole genome were plotted.
Spatiotemporal expression analyses of pseudogenes
Fastp 38 (version 0.21.0) was used to filter the original transcriptome sequencing reads, and the samples with the total clean reads number greater than or equal to 10 million were retained. We used HISAT2 39 (version 2.0.4) to align RNA-seq reads to the silkworm reference genome. The reference annotation file was obtained by merging the silkworm pseudogenes and protein-coding genes. The transcripts for each sample was assembled by stringtie 40 (version 2.2.1) with above reference annotation file, and the transcripts of all samples were merged by stringtie to obtain the non-redundant transcripts. We then extracted the transcripts of the pseudogene locus in each sample and the pseudogene gene was expressed if its TPM ⩾ 1. The criterion for judging whether a pseudogene gene was expressed in a certain developmental stage or a certain tissue was that at least 3 samples from that developmental stage or tissue had an expression level (TPM) ⩾ 1.
Differential expression analyses were performed on the following groups of spatio-temporal sample groups: (1) 3 different cell lines; (2) silk gland tissues at different developmental stages; and (3) fat body tissues at different developmental stages. For each group, the counts of pseudogenes in each sample were calculated using the prepDE.py script provided by stringtie and then converted into log2(count + 1) to construct an expression matrix. Then, the edgeR was used to analyze the differentially expressed pseudogenes in each group. Finally, the pseudogenes with |log FC| > 2 and FDR value <0.001 were extracted as differentially expressed pseudogenes (DEGs).
Tissue-specific and differential expression analyses
The average expression of pseudogenes in different tissues was calculated, and tissue-specific indicators were calculated using Tspex 41 (version 0.6.1). Then, tissue-specific genes were filtered by using different cutoff values (1, 0.95, 0.9, 0.85, and 0.8).
Identification of pseudogene-derived lncRNA genes
To identify the pseudogenes derived lncRNAs in the silkworm, we used 3 different software, CPC2 42 (version 1.0.1), LGC 43 (version 1.0), and PLEK 44 (version 1.2), combining with characteristics such as open reading frame (ORF) and K-mer. The pseudogene was defined as lncRNA candidate if it was predicted as lncRNA by at least 2 of 3 software. Then, GO enrichment analysis was performed on the parent genes of the pseudogenes that were identified as lncRNAs.
Target gene prediction of pseudogene-derived lncRNAs
We used 2 methods to predict the target genes of pseudogenes derived lncRNAs. First, we used LncTar 45 (version 2.0), which uses the base-pairings to calculate the minimum free energy of the joint structure of 2 RNA molecules, to explore the lncRNA-mRNA interaction. By inputting lncRNA and mRNA sequences, the ndG cut-off value was set to −0.13, and the possible targeted genes of pseudogenes derived lncRNAs in silkworm were predicted. Then, using the pandas (version 1.5.3) and scipy (version 1.11.1) libraries in Python (version 3.10.12), the Spearman correlation coefficients between lncRNAs and mRNAs using their TPM expression matrix. The thresholds r ⩽ −.75 and P value < .01 were set to obtain the predicted targeted genes of pseudogenes derived lncRNAs. The intersections of the above 2 methods were used as the final targeted genes of pseudogenes derived lncRNAs. We also performed GO enrichment analysis of pseudogenes derived lncRNAs targeted genes.
Results
The landscape of demotic silkworm pseudogenes
Collecting and filtering high-quality reference protein sequences is the first step in identifying pseudogenes and is the most important aspect for accurate results. Based on the high-quality reference protein sequences, we identified 4410 pseudogenes through the process illustrated in Figure 1A. The functions of parent genes have guiding significance for the functional analysis of pseudogenes. After GO enrichment analysis and filtering the results with adjusted P < .01 as the condition, we found that pseudogenes were enriched in DNA binding and NADH dehydrogenase activity (ubiquinone), nucleosome assembly, transposition (DNA-mediated), mitochondrial electron transport (NADH to ubiquinone), DNA-templated transcription (initiation) (Table 1).

Identification, classification, and distribution of pseudogenes in the silkworm. (A) the pipeline for pseudogenes identification. (B) Classification and statistics of pseudogenes. Including duplications (DUPs), unitary pseudogenes (Unitary), processed pseudogenes (retropseudogenes, RETs), and fragments (FRAGs). (C) The distribution of pseudogenes and the ratio of pseudogenes to protein-coding genes on silkworm chromosomes. The statistical unit of the distribution of pseudogenes and protein-coding genes is 100 KB, and the change of chromosome color is used to describe the ratio of the number of pseudogenes to the number of protein-coding genes in the statistical region.
GO enrichment analysis of pseudogenes’ parent genes.

These pseudogenes could be divided into 4 categories and 7 subcategories. These 4 categories were Unitary, RET, FRAG, and DUP, and the 7 subcategories were Unitary-truncated, Unitary-full, RET-truncated, RET-full, FRAG, DUP-truncated, and DUP-full. The identification results are presented in Supplemental Table S2. As shown in Figure 1B, among these 7 subcategories, Unitary-full had the largest proportion, followed by FRAG, and DUP-truncated had the smallest proportion. In addition, all 7 types appeared in the intergenic regions, while only DUP-full and unitary-full appeared in the exon regions. Distribution type included o, u, and y types. The distribution of these pseudogenes on silkworm chromosomes is shown in Figure 1C. Of the 4410 pseudogenes identified, 2781 were on chromosomes, and for the remaining pseudogenes, we could not determine the specific chromosome position because they were on genomic fragments that had not yet been assembled. There was no significant difference in the number of pseudogenes per chromosome. The lowest number of pseudogenes (59) was on chromosome 27, and the highest number of pseudogenes (137) was on chromosome 24. However, these pseudogenes were unevenly distributed on the chromosomes; various locations contained more than would be expected by chance, such as the front end of chromosome 11, the back end of chromosome 9, the middle of chromosome 1, and the whole of chromosome 28.
The evolutionary history of pseudogenes
The identity between the pseudogene and its corresponding parent gene reflects the evolutionary relationship of the sequence. In general, the identity between the pseudogene and its corresponding parent gene is inversely proportional to the divergence time. In the analysis of the identity of pseudogenes and their corresponding parental genes, we excluded the statistics of the Unitary type because Unitary pseudogenes are derived from a single-copy gene variation in this species, and the parent gene of this species as a reference has been lost. The parental identity was distributed between 0.3 and 1, of which the most common value was in the range of 0.3 to 0.4, followed by 0.5 to 0.6 and 0.4 to 0.5 (Figure 2A). In terms of types, only FRAG types appeared in the range of 0.7 to 0.8, and only DUP and FRAG types appeared in the range of 0.9 to 1(Figure 2A).

Evolutionary analyses of pseudogenes in the silkworm. (A) Identity distribution of pseudogenes and their corresponding parent genes. Including DUP-full, DUP-truncated, FRAG, RET and RET-truncated. (B) Density distribution of pseudogene divergence times in the whole genome (Mya: million years ago). (C) The Ka (Number of nonsynonymous substitutions per nonsynonymous site), Ks (Number of synonymous substitutions per synonymous site) and Ka/Ks plot of pseudogenes and their parent genes.
We then calculated the divergence time of each pseudogene and its parent gene by using the mutation rate μ = 1.3e-8 substitutions per site per year and the formula: t = (d/2)/μ to calculate the formation time of pseudogenes, and the results are shown in Figure 2B. Most of the pseudogenes were formed between 1 and 2 mya, and a large portion of the pseudogenes were formed around 25 mya, with another portion of the pseudogenes formed around 35 mya.
The results of pseudogene selection pressure analysis are shown in Figure 2C. Ka values were largely distributed between 0.2 and 0.8, while Ks values were distributed around 4. The selection pressure can be calculated by Ka and Ks via the ratio Ka/Ks. According to the calculated Ka/Ks distribution, most of the pseudogenes were negatively selected (ie, purifying selection, Ka/Ks < 1); however, some were positively selected (Ka/Ks > 1).
Global expression patterns of pseudogenes
In an effort to investigate the spatial and temporal expression patterns of pseudogenes in silkworms, we gathered RNA sequencing (RNA-Seq) data sets from over 1200 silkworm samples archived within the Sequence Read Archive (SRA) form NCBI. Following a meticulous review of these entries from the NCBI database and the relevant scholarly articles, we were able to secure 832 samples that provided clear-cut details regarding tissue types or developmental phases (Supplemental Table S1). Using the integrated genome annotation files (including protein-coding genes and pseudogenes in this study) as reference, we obtained the average TPM value of pseudogenes in all samples. According to expression levels, the identified pseudogenes were roughly divided into 2 categories: high expression and low expression (Figure 3). In general, the highly expressed genes were expressed in different periods and in different tissues, and the expression level was relatively high.

Gobble expression pattern of pseudogenes. The pseudogenes are clearly divided into 2 cluster, a highly expressed (A) and a lowly expressed (B). Different Age and tissue are marked above the figure. On the left side is the clustering of different samples. The different colors in the heat map represent the difference in expression.
GO enrichment analysis of high-expression and low-expression pseudogenes was performed and the results were filtered using adjusted P < .01. The results showed that the high-expression pseudogenes were mainly enriched in NADH dehydrogenase (ubiquinone) activity and Cytochrome-c oxidase activity, while the low-expression pseudogenes were mainly enriched in DNA binding, Nucleosome assembly, DNA transposition and Nucleus (Table 2). However, some pseudogenes with low overall expression seemed to show certain tissue specificity or age specificity, a result that deserves further exploration.
GO enrichment analysis of HE and LE pseudogenes’ parent genes.

HE indicates high expression, LE indicates low expression.
Temporal and spatial expression patterns
To further explore the temporal and spatial specificity, we determined whether the pseudogenes were expressed in all samples under the condition of TPM > 1. Afterward, we analyzed the expression of pseudogenes in different developmental stages and in different tissues. The results are shown in Figure 4A and B.

Expression profiling of pseudogenes in different developmental stages (A) and tissues (B). The pseudogenes are expressed its TPM > 1. The barplot at the left is the number of pseudogenes in each tissue or developmental stages, while the barplot at the top is the number pseudogenes for each combination of tissue or developmental stage. The single point in the graph represents the type-specific pseudogenes, and the multiple points and the connections between them represent the same pseudogenes in the type they cover.
As shown in Figure 4A, age-specific pseudogenes were found in 8 periods, except for 2nd_larva. In addition, 98 pseudogenes were co-expressed in the larval stage, 662 pseudogenes were co-expressed in both the prepupal and pupal stages, and 779 pseudogenes were co-expressed in both eggs and in vitro cells. In addition to the period-specific pseudogenes, we also found 92 pseudogenes that were co-expressed in all of our survey periods.
Figure 4B roughly shows the expression patterns of pseudogenes in different tissues. The results showed that there were a number of specifically expressed pseudogenes in each tissue type listed. In addition, we also found 467 co-expressed pseudogenes in embryonic tissue samples and cells derived from embryos. Then, 226 co-expressed pseudogenes were found in fat body and isolated fat body cells, and 384 co-expressed pseudogenes were found in the ovary tissue and its derived in vitro cells (BmE). In addition, 258 co-expressed pseudogenes were found in different parts of the silk gland. Finally, we found that 81 pseudogenes were expressed in all tissues.
Tissue-specific expression of pseudogenes
According to the global and tissue-specific expression patterns of the pseudogenes, we speculated that there may be some tissue-specific pseudogenes with important functions. The tissue-specific index (TSI) value is an important criterion for measuring the degree of tissue-specific expression of a gene. TSI can be calculated using Tpsex software, and its value range is 0 to 1; the closer the value to 1, the higher the degree of tissue specificity. Based on the log10 (TPM) of pseudogenes in different tissues, the TSI values of pseudogenes in each tissue were calculated using Tpsex (Supplemental Table S2). Subsequently, we used different thresholds to analyze the results, and we determined whether the expression of these pseudogenes was tissue-specific and which tissues were involved. The results are shown in Figure 5.

Tissue-specific expression of silkworm pseudogenes. The bar plots are the number of tissue-specific expressed silkworm pseudogenes, and also present on the diagram. The TSI is set to a maximum value of 1, which represents the pseudogene with the most tissue-specific expression characteristics.
As shown in Figure 5, with the TSI threshold set to 1.0, we found a total of 547 pseudogenes with tissue-specific expression characteristics. The tissue with the most tissue-specific pseudogenes was the posterior silk gland, followed by the midgut and BmN cells that derive from the ovary. In the trachea, anterior silk gland, and pupal cuticle, only 1 tissue-specific pseudogene was found. No tissue-specific pseudogenes were found in the other 10 examined tissues.
Differential expression of pseudogenes in cells, silk gland, and fat body
For differentially expressed pseudogenes, we selected 3 tissues, isolated cells, silk glands, and fat bodies. Silkworm isolated cells are important materials for studying gene function, and silk gland and fat body are closely related to the economically important traits and the health of the silkworm in agricultural production. According to the results of the expression level analysis, the edgeR program was used to analyze the differentially expressed genes in each group, and the pseudogenes with the final filter |log FC| ⩾ 2 and FDR < 0.001 were set as differentially expressed genes.
As shown in Table 3, the number of differentially expressed genes between embryo BmE cells and ovary BmN4 cells ranked first at 728. The number of differentially expressed genes between different parts of the silk gland ranged from 196 to 1, with an average of 81.8. In different stages of the fat body, the highest number of differentially expressed pseudogenes was 153 (moth vs pre pupa), while the least was 2 (fifth instar vs wandering stages), with an average of 49.5. Some of the differentially expressed genes were not enriched in a certain function, and other differentially expressed pseudogenes that could be enriched to functions were enriched in “DNA integration.” In addition, in vitro E cells and N cells were also enriched in the “transposition, DNA-mediated function.”
The number of differentially expressed pseudogenes in silkworm under different developmental stages and tissues.

Identification and analyses of lncRNAs derived from pseudogenes
The identification results of lncRNAs in the pseudogenes of the silkworm showed that there were 3299 candidate lncRNAs, accounting for 0.75 (3299/4410) of the total. Subsequently, the scope was further narrowed according to the prediction results of the target genes of lncRNAs. Among these, 776 lncRNAs had predicted target genes, accounting for 0.176 (776/4410) of the total number of pseudogenes.
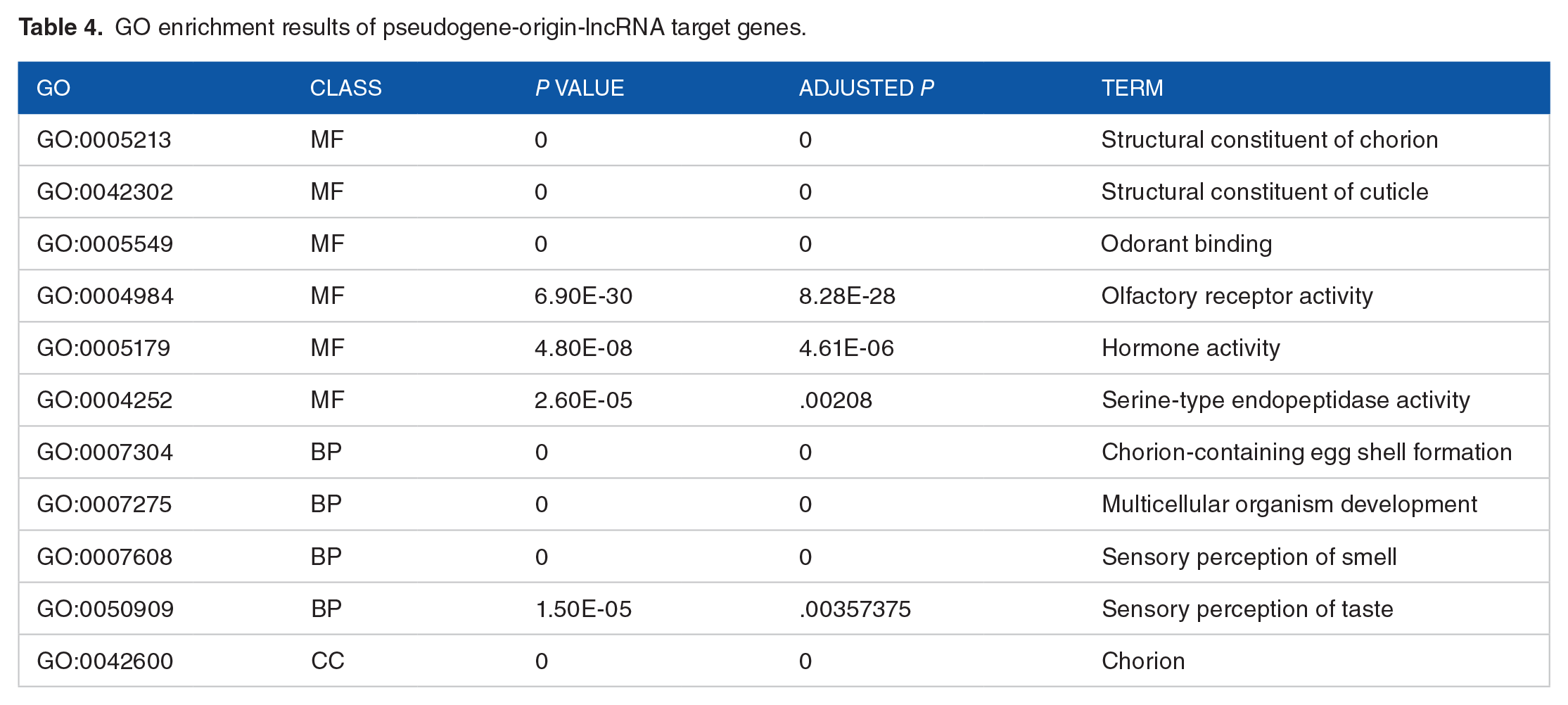
We performed GO enrichment analysis of the parent genes of lncRNAs derived from pseudogenes (Table 4 and Supplemental Table S3). We used an adjusted P value < .01 as the screening condition. Compared with the GO enrichment results for the parent genes of the pseudogenes, the enrichment results were basically the same, with only the nucleus (CC) added. GO enrichment analysis of the target genes of lncRNAs showed that these lncRNAs were enriched in MF, BP, and CC. There are 2 points worth noting in all GO enrichment results. First, these target genes were highly correlated with the eggshell formation process; second, they were also highly correlated with olfaction.
GO enrichment results of pseudogene-origin-lncRNA target genes.
Discussion
In this study, we used the genome assemblies of the silkworm and close related species and constructed a high-confidence protein set for pseudogene identification. Based on these datasets, there were 4410 pseudogenes in the silkworm (genome size: 460 Mbp), accounting for 0.27 of the total protein-coding genes in the silkworm (16 069). The ratio of pseudogene to protein-coding genes in Drosophila is much smaller 0.097 (1371/14 076). According to the NCBI database, the number of pseudogenes identified in humans (genome size: 3.1 GB) is 17 487, accounting for 0.85 of the total protein-coding genes (20 653). The total number of pseudogenes identified in mice (genome size: 2.7 GB) is 11 404, accounting for 0.43 of the total protein-coding genes (26 341). Prade et al 32 conducted an in-depth study of the numbers of pseudogenes and found that the pattern was primarily determined by the generation rate and extinction rate of pseudogenes in each species. The ratio of pseudogene to protein-coding genes in Drosophila and B. mori are lower than those in humans and mice. A possible reason is that the smaller genome and fewer protein-coding genes reduce the probability of pseudogene generation. In addition, the generation times of Drosophila and B. mori are shorter than human mice, resulting in more rapid population turnover and hence removal of pseudogenes.
In our study, we found that the divergence time of pseudogenes and their parent protein-coding genes were concentrated at 25 to 35 million year ago. The Anticla family is the closest to Bombycidae, and their divergence time is about 48.9 to 50.9 mya, 46 indicating these pseudogenes were generated after their divergence. Considering that the divergence time between the wild silkworm and the domestic silkworm is about 0.005 mya, 15 the most of pseudogenes were generated before their divergence and may be the manifestation of genus. In addition to the above peaks, there was also a concentration near 0 mya, possibly due to the domestication of silkworms by humans 5000 years ago along with selection for their agronomic traits and the “tolerance of variation,” which eventually led to the gradual formation of 2 species.
Whether a sequence can be transcribed into mRNA is an important criterion for pseudogenes to be able to perform their functions. To study the spatial and temporal expression patterns of pseudogenes in silkworms, we have meticulously selected 832 samples with precise and clear tissue and developmental stage information (Supplemental Table S1). We found that these pseudogenes were clearly divided into 2 cluster, a highly expressed and a lowly expressed (Figure 3). The analyses of pseudogenes expression in different developmental stages and in different tissues revealed that 92 pseudogenes were co-expressed in all of our survey periods and 81 pseudogenes were expressed in all tissues (Figure 4). All these highly and widely expressed pseudogenes may be curial for silkworm development. Totally, 547 pseudogenes with tissue-specific expression characteristics were identified. Interestingly, the posterior silk gland, an organ that determines the economic traits of the silkworm, was the tissue with the most tissue-specific pseudogenes (199), which is consistent well with the factor that the silkgland is highly specialized tissues, and implying these pseudogenes may be involved in the development and function of silkgland in the silkworm.
With the improvement in research methods, especially the development of bioinformatics and high-throughput sequencing technology, researchers have begun to realize that pseudogenes may have important biological significance.2,6 Some pseudogenes can encode proteins that are shorter than the parental functional genes but do have functions. 7 Some pseudogenes regulate gene expression through transcription into RNA-mediated regulatory species such as lncRNA, siRNAs (Small interfering RNA), miRNA (MicroRNA), and snoRNA (Small nucleolar RNA). 8 Here, we identified 3299 candidate pseudogene-sourced lncRNAs, accounting for 0.75 of the total pseudogenes (4410), implying a dominant role for pseudogenes in the silkworm.
Totally, 776 lncRNAs had predicted target genes, accounting for 0.176 of the total pseudogenes. Based on the GO enrichment results of parent genes and target genes of pseudogene-derived lncRNAs, it appears that lncRNAs do not frequently appear in GO terms that have a significant impact on the survival and reproduction of organisms. Additionally, the target genes of lncRNAs do not seem to have a significant effect on the survival and reproduction of organisms under normal circumstances. From this, we can speculate that genes that play crucial roles in the survival and reproduction of organisms may have detrimental effects on the competitiveness of organisms if they become pseudogenes. In other words, if these genes were to become “non-functional” pseudogenes, this could negatively affect the organism’s ability to compete with conspecifics or other species. Furthermore, it is possible that organisms can survive normally before such pseudogenes are generated. However, if the target genes of these lncRNAs are important for the survival and reproduction of organisms and are negatively regulated by the lncRNAs, this could also have a significant negative impact on environmental competitiveness. In the course of evolution, organisms that possess these “non-functional” pseudogenes that are negatively regulated by lncRNAs may have existed but eventually disappeared. This suggests that the presence of such pseudogenes and their regulation may have led to the extinction of certain organisms over time.
Our study provided the systematic analyses of pseudogenes in the silkworm, including the identification in genome wide, the evolutionary history, the spatio-temporal expression profiles, and their biological roles as lncRNAs. However, there also have some limitations. First, pseudogenes involve in many biological processes via lncRNAs, siRNAs (Small interfering RNA), miRNA (MicroRNA), snoRNA (Small nucleolar RNA), 8 truncated proteins 7 etc., however, here we only explored the functional roles of pseudogenes as lncRNAs, and more potential roles of pseudogenes remain to be studied further. Second, the silkworm pangenome project have provided population level genomic variations, however, whether the pseudogenic loci have covered some of these variations and these variations located in pseudogenes are associated to traits need to be explored.
Conclusions
Pseudogenes are significant in regulating gene expression, and analysis of pseudogenes can shed light on species evolution. Through the genome data of silkworm and related species, we identified 4410 pseudogenes of the silkworm and constructed a pseudogene expression map to analyze the expression characteristics of pseudogenes through 832 silkworm transcriptome datasets. Among these pseudogenes. We identified 3,299 lncRNAs in these pseudogenes, and 776 lncRNAs and their target genes were identified. the target genes of these lncRNAs in silkworm pseudogenes were enriched in the egg formation and olfactory function. Overall, this study provides valuable insights into the regulation and expression characteristics of pseudogenes in the silkworm. It also has the potential to contribute to the study of pseudogenes in other organisms.
Supplemental Material
sj-xlsx-1-evb-10.1177_11769343241261814 – Supplemental material for The Spatio-Temporal Expression Profiles of Silkworm Pseudogenes Provide Valuable Insights into Their Biological Roles
Supplemental material, sj-xlsx-1-evb-10.1177_11769343241261814 for The Spatio-Temporal Expression Profiles of Silkworm Pseudogenes Provide Valuable Insights into Their Biological Roles by Linrong Wan, Siyuan Su, Jinyun Liu, Bangxing Zou, Yaming Jiang, Beibei Jiao, Shaokuan Tang, Youhong Zhang, Cao Deng and Wenfu Xiao in Evolutionary Bioinformatics
Supplemental Material
sj-xlsx-2-evb-10.1177_11769343241261814 – Supplemental material for The Spatio-Temporal Expression Profiles of Silkworm Pseudogenes Provide Valuable Insights into Their Biological Roles
Supplemental material, sj-xlsx-2-evb-10.1177_11769343241261814 for The Spatio-Temporal Expression Profiles of Silkworm Pseudogenes Provide Valuable Insights into Their Biological Roles by Linrong Wan, Siyuan Su, Jinyun Liu, Bangxing Zou, Yaming Jiang, Beibei Jiao, Shaokuan Tang, Youhong Zhang, Cao Deng and Wenfu Xiao in Evolutionary Bioinformatics
Supplemental Material
sj-xlsx-3-evb-10.1177_11769343241261814 – Supplemental material for The Spatio-Temporal Expression Profiles of Silkworm Pseudogenes Provide Valuable Insights into Their Biological Roles
Supplemental material, sj-xlsx-3-evb-10.1177_11769343241261814 for The Spatio-Temporal Expression Profiles of Silkworm Pseudogenes Provide Valuable Insights into Their Biological Roles by Linrong Wan, Siyuan Su, Jinyun Liu, Bangxing Zou, Yaming Jiang, Beibei Jiao, Shaokuan Tang, Youhong Zhang, Cao Deng and Wenfu Xiao in Evolutionary Bioinformatics
Supplemental Material
sj-zip-4-evb-10.1177_11769343241261814 – Supplemental material for The Spatio-Temporal Expression Profiles of Silkworm Pseudogenes Provide Valuable Insights into Their Biological Roles
Supplemental material, sj-zip-4-evb-10.1177_11769343241261814 for The Spatio-Temporal Expression Profiles of Silkworm Pseudogenes Provide Valuable Insights into Their Biological Roles by Linrong Wan, Siyuan Su, Jinyun Liu, Bangxing Zou, Yaming Jiang, Beibei Jiao, Shaokuan Tang, Youhong Zhang, Cao Deng and Wenfu Xiao in Evolutionary Bioinformatics
Footnotes
Acknowledgements
We would like to thank the previous researchers for providing genome data of the silkworm and related species and transcriptome data from massive silkworm samples in the NCBI (National Center for Biotechnology Information) database. We Thank Prof. Jinshu Xiao and Master Bowen Yin for their help in the revision of the paper. We thank letpub (![]() ) for its linguistic assistance during the preparation of this manuscript.
) for its linguistic assistance during the preparation of this manuscript.
List of abbreviations
DUP: duplications
Unitary: unitary pseudogenes
RET: processed pseudogenes (retropseudogenes)
FRAG: fragments
lncRNA: long non-coding RNAs
Authors’ Contributions
WFX and CD conceived and designed the study. LRW, SYS prepared the original manuscript. LRW, SYS, JYL, BXZ, YMJ, BBJ, and SKT performed bioinformatics analysis. YHZ, WFX and CD reviewed and edited the manuscript. LRW, JYL, and BBJ helped organize the figures. All authors contributed to the article and approved the submitted version.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the China Agriculture Research System project (NO. CARS-18), the Sichuan Province “14th 5-Year Plan” crop and livestock breeding project (NO. 2021YFYZ0024), Aquatic (silkworm) livestock and poultry breeding project (NO. 1+9KJGG004), Nanchong applied Basic Research Project (NO. 23YYJCYJ0044) and Silkworm artificial feed adaptive resource creation and application project (NO. 2022ZZCX084). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Approval and Consent to Participate
No animal studies are presented in this manuscript. No human studies are presented in this manuscript. No potentially identifiable human images or data are presented in this study.
Consent for Publication
Not applicable.
Availability of Data and Materials
Not applicable.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.