
Abstract
In the present paper we review the translocation network involving TET and ETS family members with special focus on the Ewing family of tumors. FUS (fusion, involved in t(12;16) in malignant liposarcoma = TLS,
Introduction
Several tumor entities are characterized by recurrent chromosomal aberrations which lead to the activation of oncogenes. Two types of oncogene activation can be distinguished: Firstly, regulatory elements of a gene can be juxtaposed to an oncogene, resulting in loss of physiological regulation of this oncogene. A well known example for this type of oncogene activation is Burkitt's lymphoma (BL). The majority of BL carry translocations between the MYC (v-myc myelocytomatosis viral oncogene homolog) oncogene and one of the immunoglobulin loci.
1
High expression of MYC in BL is driven by regulatory elements of the immunoglobulin loci
2
and deregulated expression of MYC in B cells is a major factor for the malignant phenotype of BL cells.3–5 Formation of tumor specific fusion proteins represents the second type of oncogene activation. In the present paper, we focus on fusion proteins of the TET-ETS (
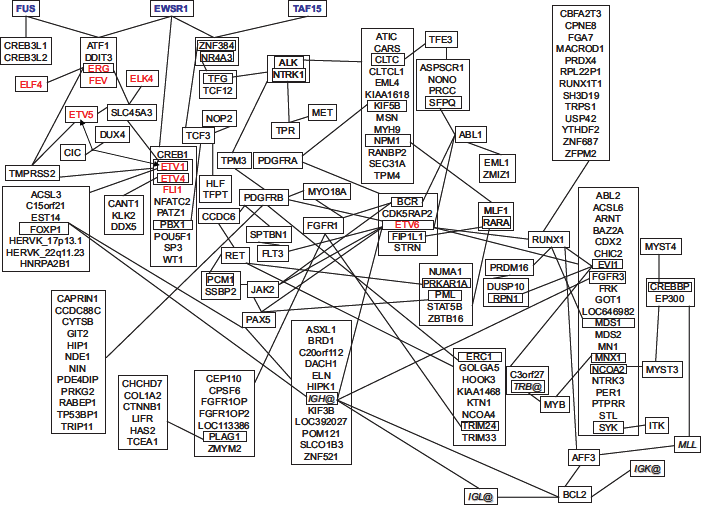
This class of oncogenes has been described in Ewing family tumors (EFT). Initially described as endothelioma, 6 EFT represent a group of bone and soft tissue sarcomas with uncertain histogenetic origin. Gene expression analyses indicate a relationship between EFT and endothelial, neuroectodermal, as well as mesenchymal stem cells.7–10 The majority of EFT carry chromosomal translocations between chromosomes 11 and 22. 11 By molecular analysis of this translocation a gene fusion between EWSR1 (Ewing sarcoma breakpoint region 1) and FLI1 (Friend leukemia virus integration 1) was detected. 12 EWSR1-FLI1 is the proto-type of fusion proteins involving members of the TET family of RNA binding proteins and members of the ETS family of transcription factors. In addition to TET-ETS fusions, translocations between TET genes and other fusion partners have been identified (Fig. 1 and Table 1).13–41 ETS transcription factors can be divided into several groups and sub-families. 42 All ETS transcription factors that have been identified as fusion partners for TET proteins are members of the sub-family ETS and are included in the groups PEA3 or ERG (PEA3 group: ETS variant 1 (ETV1), ETS variant 4 (ETV4 = PEA3, polyomavirus enhancer activator-3); ERG group: ETS related gene (ERG), fifth Ewing variant (FEV), FLI1). A third gene in the PEA3 group, ETV5 (ETS variant 5) has not been identified as fusion partner for TET proteins. However, ETV5 is up-regulated (together with ETV1) by CIC-DUX4 (capicua homolog-double homeobox, 4) oncofusion proteins which have been found in so called “Ewing-like sarcomas”. 43 Gene fusions between ETV1, ETV4, ETV5 or ERG and different other fusion partners have also been detected in prostate cancer.43–51 One of these fusion partners, SLC45A3 (solute carrier family 45, member 3), forms additional gene fusions with the ETS family member ELK4 (ETS-like transcription factor 4). 52 Like the other fusion partners, ELK4 is a member of the sub-family ETS, but represents the first member of the ELK group. 42 An unusual gene fusion between two ETS family members has been described in acute myeloid leukemia. 53 This fusion leads to the formation of fusion proteins between ERG and ELF4 (E74-like factor 4), a member of the ELF sub-family of ETS transcription factors.

Gene fusions involving members of the TET family.

Material and Methods
Cell line SK-N-MC 54 was obtained from the Deutsche Sammlung für Mikroorganismen und Zellkulturen (Braunschweig, Germany). Cells were cultured in RPMI1640 medium supplemented with 10% fetal calf serum and penicillin/streptomycin. For visualization of mitotic figures in living cells, cells were transfected with vector pBOS-H2BGFP55,56 (Becton-Dickinson, Heidelberg, Germany) and stable transfectants were selected by treatment with 2 µg/ml blasticidin. Information about gene fusions was collected from the literature and the Mitelman Database of Chromosome Aberrations in Cancer. 57 For identification of EWSR1 pseudogenes we performed a BLAST search 58 using the mRNA sequence of EWSR1 exons 1–7 as query. For comparison of EWSR1 transcripts and pseudogenes, the open reading frame of EWSR1-FLI1 type I was amplified from cell line A-67359 with EWSR1 and FLI1 specific primers (5′-TTG GAT CCG CTT CAG CTA GAA GGC CAC T-3′; 5′-AAA AGC TTA TGG CGT CCA CGG ATT AC-3′) and sequenced by using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) according to manufacturer's instructions. Sequence alignment between the EWSR1 gene, EWSR1 transcripts and pseudogenes was visualized by using GeneDoc. 60
Results and Discussion
Several of the fusion partners of TET and ETS family members are involved in additional chromosomal rearrangements. For example, PBX1 (pre-B-cell leukemia homeobox 1) and ZNF384 (zinc finger protein 384) are both involved in translocations with EWSR1 and the unrelated transcription factor TCF3 (transcription factor 3).61,62 The observation that such translocations often occur in tumors of the same type as the corresponding translocations involving TET and/or ETS family members suggest that these translocations have similar pathophysiological effects. For instance, fusion proteins between the TET proteins EWSR1 or TAF15 and NR4A3 (nuclear receptor subfamily 4, group A, member 3; a member of the steroid/thyroid hormone and retinoid receptor super-family) 63 have been observed in myxoid chondrosarcoma.23,41 In the same tumor type translocations between NR4A3 and TCF12 (transcription factor 12) 64 or TFG (neurotrophic tyrosine kinase, receptor, type 1-fused gene) 65 have been found. Interestingly, TFG was found initially by searching for TET family members, 66 but (like TCF12) has only low sequence similarity with EWSR1, FUS and TAF15. Such rearrangements link TET-ETS translocations with seemingly unrelated gene fusions, including fusions involving the ETS transcription factor ETV6 (ETS variant 6; Fig. 2). ETV6 (also known as TEL: translocation, ETS, leukemia) is the only ETS family member for which gene fusions have been described but which is not involved in gene fusions with TET family members or SLC45A3 (solute carrier family 45, member 3). ACSL3 (acyl-CoA synthetase-like 3)-ETV150 and ETV6-ACSL6 (acyl-CoA synthetase-like 6) 67 gene fusions both involve an ETS family member and a member of the long chain acyl-CoA synthetase family. However, whereas ASCL3-ETV1 gene fusions allow the expression of a truncated ETV1, 50 it seems that fusions between ASCL6 and ETV6 did not allow expression of ETV6 or fusion proteins. Therefore, these two gene fusions might exert different pathogenetic functions.
The formation of fusion genes involving members of the TET and/or ETS family has multiple oncogenic effects. One effect is the deregulated expression of the 3′ fusion partner which is driven by the promoter of the 5′ fusion partner. In prostate cancer, the formation of fusion proteins has been described, 46 but most gene fusions lead to up-regulation of ETS-factors by heterologous promoters without formation of fusion proteins.44,50 Aberrant regulation of target genes can explain some of the oncogenic activity of gene fusions involving ETS family members or other transcription factors. The EFT specific EWSR1-FLI1 oncofusion protein has similar DNA binding specificity as FLI1. 68 In addition, up-regulation of ETS factors by unrelated oncogenic events in so called “Ewing-like sarcomas” suggest that activation of ETS factors without formation of novel fusion proteins is largely sufficient for induction of the EFT phenotype. 43

On the other hand, EWSR1-FLI1 has a higher transformation activity than FLI1,
69
indicating that the fusion protein has oncogenic capacities that are not attributed to over-expression of the ETS translocation partner alone. Recent evidence indicate that the chimeric EWSR1-FLI1 transcription factor bind not only ETS consensus sites but also microsatellite sequences. The enrichment of such sequences in proximity to known EFT associated genes suggests that EWSR1-FLI1 fusion proteins regulate gene expression after binding to these microsatellite sequences.
70
In addition to the direct regulation of gene expression, EWSR1-FLI1 exert transcription factor activity-independent oncogenic activities. EWSR1-FLI1 activates gene transcription,70,71 but tumorigenicity of EWSR1-FLI1 is partially independent on DNA binding.
72
Interference of several TET fusion proteins with the splicing machinery has been observed.73–76 Because TET proteins are involved in RNA splicing,77–79 this might be indicative for an interference between wild type TET proteins and fusion proteins. Indeed, inhibition of wild type EWSR1 function by EWSR1-FLI has been described.
80
Interestingly, this inhibition leads to mitotic defects. Aberrant mitotic figures can be regularly observed in cultured EFT cells (Fig. 3) and might be responsible for the high frequency of secondary chromosomal aberrations seen in EFT.81,82 Finally, the different activities of the fusion proteins lead to altered gene expression. Despite a similar histological appearance, TET-ETS positive EFT have a gene expression profile which clearly discriminates these tumors from other tumors of the family of so called small round blue cell tumors.7,83,84 Several target genes of TET-ETS fusion proteins have been identified85–99 and different TET-ETS fusion proteins induce a similar tumorigenic activity in transgenic cells.
100
On the other hand, the consequences of TET-ETS fusion protein activity depend on the cellular background101,102 and fusion type dependent differences in the gene expression profile of EFT have been observed.103,104 The fusion type as well as the genes expression profile have been identified as prognostic factors for EFT patients.105–109 The exact function of TET-ETS fusion proteins in cancer pathogenesis remains unclear. Case reports describing patients with two TET-ETS translocations in the same tumor might indicate that TET-ETS translocations are not the primary event leading to tumor formation.
110
Nevertheless, TET-ETS fusion proteins are required for growth of EFT and several strategies for the inactivation of TET-ETS fusion transcripts have been developed.111–114 EFT have a neuronal phenotype which can partially be explained by the activity of EWSR1-ETS fusion proteins.7,115–118 TET proteins are involved in neuronal biology and a link between TET proteins and neurodegenerative diseases have been established.119,120 However, the function of wild type TET proteins is not restricted to the nervous system but is also required for hematopoiesis.
121
Surprisingly, EWSR1-FLI1 expressing transgenic animals did not develop EFT like sarcomas but leukemia.
122
Restricted expression of the fusion proteins in mesenchymal cells leads to sarcoma formation only in the setting of additional TP53 aberrations.
123
These observations indicate that TET-ETS fusion proteins are not sufficient to induce tumor formation. Similarly, TET-ETS fusion proteins are not sufficient to induce the complete gene expression program of EFT. Which factors are responsible for expression of EFT-associated but TET-ETS-independent genes (e.g. lipase member I, LIPI)
124
is unknown. Recently, tumor stem cells in EFT have been identified.
125
Interestingly, these cells are characterized by expression of transcription factors NANOG (Tir Na Nog) and POU5F1 (POU (Pituitary-specific 1,

Recent evidence indicates that epigenetic mechanisms play a major role in cancer pathogenesis mediated by TET and/or ETS gene fusions. Epigenetic inactivation of tumor suppressor genes have been observed in EFT 129 and inhibitors of histone deacetylation or DNA methylation have been shown to exert anti-tumor activity against EFT.130,131 Similarly, TMPRSS2 (transmembrane protease, serine 2)-ETS translocations in prostate cancer are associated with increased histone deactetylase expression. 132 One of the target genes of EWSR1-FLI1, enhancer of zeste homolog 2 (EZH2), is involved in epigenetic inactivation of genes. Based on the observation of high expression of EZH2 in EFT 7 a model for epigenetic inactivation of differentiation inducing genes was proposed.10,133 This model implicates that up-regulation of epigenetic silencers like EZH2 by TET-ETS fusion proteins fix the tumor cell in an un-differentiated state. In deed, inhibition of EZH2 allows differentiation of EFT cells and inhibits tumor growth. 10 Another implication of this model is that different primary oncogenic events might be fixed by TET-ETS fusion proteins leading to similar tumor morphology.
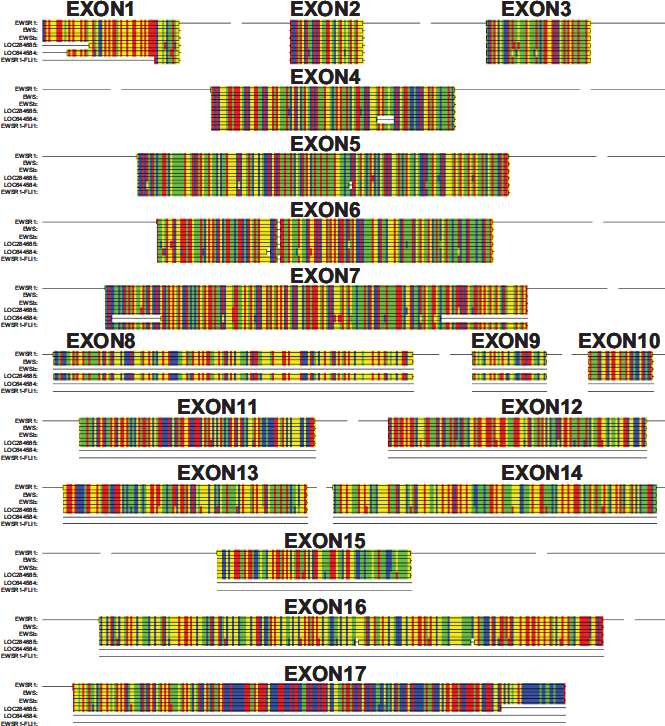
Today, only three members of the TET family have been identified in the human genome. Another RNA binding protein, RBM14 (RNA binding motif 14), with weak similarity to TET proteins and involvement in regulation of RNA transcription and splicing has been identified, 134 but translocations involving this gene have not been found. Interestingly, the human genome contains EWSR1 pseudogenes. 135 By using the cDNA sequence of the EWSR1 part from the EWSR1-FLI1 type I translocation as bait we found two human pseudogenes. The presence of multiple EWSR1 pseudogenes in the human genome indicates that EWSR1 sequences have repeatedly been involved in rearrangements. One pseudogene on chromosome 1 (LOC284685) contains an intronless copy of the complete open reading frame of EWSR1 corresponding to the longer (EWS) isoform (Fig. 4). Several point mutations did not allow the translation of a corresponding EWSR1 protein. The second copy on chromosome 14 (LOC644584) is again an intronless copy of the open reading frame of EWSR1. However, this copy contains only the 5′ part (corresponding to exons 1 to 7) of EWSR1 and resembles EWSR1 in the most common type 1 EWSR1-FL1 translocation (Fig. 4). Again, several point mutations and deletions did not allow translation of a protein. In this pseudogene the 3′ part of EWSR1 is replaced by a non-coding sequence from chromosome 3 (data not shown).
Taken together, gene rearrangements involving members of the TET or ETS families are part of a large network of oncogenic gene fusions. Whether these gene fusions can be targeted with clinical therapeutic benefit has to be shown.
Disclosures
The authors report no conflicts of interest.
Footnotes
Acknowledgements
We thank Ines Volkmer and Siggi Heins for helpful experimental assistance.