
Abstract
Corynebacterium striatum is a Gram-positive bacterium that is straight or slightly curved and non-spore-forming. Although it was originally believed to be a part of the normal microbiome of human skin, a growing number of studies have identified it as a cause of various chronic diseases, bacteremia, and respiratory infections. However, despite its increasing importance as a pathogen, the genetic characteristics of the pathogen population, such as genomic characteristics and differences, the types of resistance genes and virulence factors carried by the pathogen and their distribution in the population are poorly understood. To address these knowledge gaps, we conducted a pan-genomic analysis of 314 strains of C. striatum isolated from various tissues and geographic locations. Our analysis revealed that C. striatum has an open pan-genome, comprising 5692 gene families, including 1845 core gene families, 2362 accessory gene families, and 1485 unique gene families. We also found that C. striatum exhibits a high degree of diversity across different sources, but strains isolated from skin tissue are more conserved. Furthermore, we identified 53 drug resistance genes and 42 virulence factors by comparing the strains to the drug resistance gene database (CARD) and the pathogen virulence factor database (VFDB), respectively. We found that these genes and factors are widely distributed among C. striatum, with 77.7% of strains carrying 2 or more resistance genes and displaying primary resistance to aminoglycosides, tetracyclines, lincomycin, macrolides, and streptomycin. The virulence factors are primarily associated with pathogen survival within the host, iron uptake, pili, and early biofilm formation. In summary, our study provides insights into the population diversity, resistance genes, and virulence factors ofC. striatum from different sources. Our findings could inform future research and clinical practices in the diagnosis, prevention, and treatment of C. striatum-associated diseases.
Keywords
Introduction
Corynebacterium striatum is a microbe that typically colonizes human and animal skin, nasal passages, throats, and other body parts. Historically, until 1980, it was considered a normal microbiome and non-pathogenic. 1 However, with advances in clinical research, C. striatum has been found to cause various infections, including bacteremia, respiratory tract infections, skin infections, and nervous system infections,2 -5 there has been a significant increase in the number of reports of C. striatum causing different types of infections, and most clinical isolates are multi-drug resistant, posing challenges for clinical diagnosis and treatment. 6
C. striatum has evolved resistance to different antibiotics due to antibiotic misuse. Recent studies suggest that the antibiotic resistance of C. striatum is mainly acquired through genetic mutations and mobile genetic elements. 7 Under the selective pressure of antibiotics, C. striatum can obtain antibiotic resistance through gene mutations, such as gyrA gene mutation that results in resistance to fluoroquinolones and pgsA2 gene mutation that leads to resistance to daptomycin.8 -11 Mobile genetic elements, such as transposons, insertion sequences, and plasmids, are the main ways for C. striatum to acquire resistance genes. For instance, the pTP10 plasmid of it carries the ermX, cmx, strA, and strB genes, which confer resistance to aminoglycosides and macrolides.12 -15
Most nosocomial infections caused by C. striatum result from invasive infections due to its colonization on various medical devices before transmission to the host.4,16 -19 Studies have found that C. striatum can form biofilm on the surface of polyurethane and silicone catheters and adhere to human cells.20 -24 Moreover, it can cause death in Caenorhabditis elegans. 9 Currently, there is a substantial body of research on C. striatum infection, which primarily focuses on its resistance to drugs and resistance mechanisms.
A pan-genome refers to the complete set of genes present in all strains of a particular species, and can be categorized into 3 types: (1) core genome, which consists of genes present in all strains, (2) accessory genome, which consists of genes present in 2 or more strains but not in all strains, and (3) unique genome, which consists of genes present exclusively in one strain. 25 Pan-genomic analysis provides insights into the genome structure and diversity of pathogenic bacteria, including genome size and gene number, and is therefore of great significance for understanding their evolutionary history and transmission routes. 26 Moreover, it enables identification and analysis of virulence factors and resistance genes, such as genes encoding toxins, proteases, cell attachment molecules, outer membrane proteins, and others, which are closely associated with the pathogenicity of these bacteria. This information is essential for studying the pathogenic mechanisms of pathogenic bacteria and developing new vaccines and therapeutic methods. 27
However, there are few studies on the pan-genomic analysis of C. striatum to investigate its population diversity and evolutionary relationships, as well as the distribution of drug resistance genes and virulence factors within its population. Only one study 28 has analyzed 30 genomes of C. striatum, but the limited number of strains used in that study does not provide a representative sample of the entire population. Therefore, this study obtained whole-genome sequencing data and phenotypic information for all available C. striatum strains from public databases. After filtering and assembling the genomic data, we analyzed 314 C. striatum genomes from various geographic locations and isolated tissues. Our findings indicate that C. striatum has an open pan-genome with high intraspecific diversity. Furthermore, we identified multiple resistance genes and virulence factors through comparison with existing databases.
Materials and Methods
Data collection
A total of 298 genome sequences and 72 Whole genome sequencing (WGS) data of C. striatum were selected for this study from the National Center for Biotechnology Information (NCBI) database (ftp://ftp.ncbi.nlm.nih.gov), based on studies by other reaches.13,28 -36 The phenotypic information of each strain was obtained by searching the NCBI BioSample database and papers, such as host, geographical locations, isolated tissue, disease, etc. In order to ensure the availability of data, we only retained strains with known geographical locations and isolation tissues, and removed 14 strains with incomplete information.
Genome assembly and filter
We performed quality control on the 72 WGS data of C. striatum using Fastp 37 (default parameter). Adapter sequences were removed, and only reads with Phred values ⩾20 were retained for subsequent analysis. The retained reads were subjected to de novo assembly using SPAdes 38 (—isolate), and the resulting genomes were annotated using Prokka 39 (—kingdom Bacteria).
To evaluate the integrity of the genome sequences and proteome sequences, we used BUSCO 40 and its companion conserved gene database corynebacteriales_odb10 (default parameter), only retained the genomes with an integrity score of ⩾95%. We then used the Pyani 41 tool (-m ANIm -g –write_excel) to perform average nucleotide identity (ANI) analysis between the remaining genomes and the strains that were removed due to low alignment coverage (<80%) or alignment identity (<95%).
Pan-genomic analysis of Corynebacterium striatum
The pan-genomic features of C. striatum were inferred using Bacterial Pan Genome Analysis tool (BPGA) tools 42 (default parameter). To construct the pan-genome, PIRATE tools 43 were applied to the 314 genomes of C. striatum. To cluster genes into gene families within each genome, PIRATE utilizes machine learning to select the most appropriate identity threshold within the given range and CD-HIT 44 for clustering. Therefore, we employed the recommended parameters and utilized the recommended range of amino acid percentage identification thresholds (50%, 60%, 70%, 80%, 90%, 95%, and 98%) suggested by the software. Functional annotation of the gene families obtained by PIRATE tools was performed using the KEGG database. 45
Intraspecific diversity analysis
The differences among individual genomes, as reflected by the variations in individual non-core genes (accessory and exclusive genes) and single nucleotide variants (SNVs) within a population, can provide insights into the diversity of the population. To investigate the intraspecific diversity of C. striatum, we counted the number of non-core genes and genome-wide SNVs in each strain. The strains were grouped according to their isolation source and geographic location. The number of non-core genes was determined through analysis of gene families obtained from PIRATE, while the number of SNVs in the whole genome was obtained by comparing the genomes of C. striatum with NUCMER 46 (–maxmatch -c 100) against the reference genome C. striatum 216, followed by statistical analysis using SYRI 47 (default parameter).
To test for significant differences between the number of non-core genes and the number of SNVs in each group, we employed the method of Analysis of Variance (ANOVA). 48 The calculation formula for this analysis is as follows:
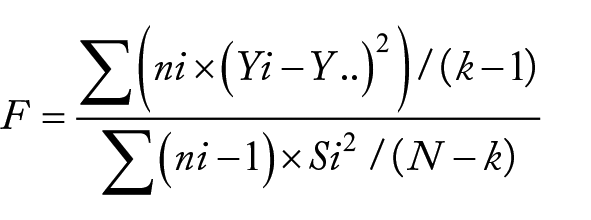
Where ni represents the sample size of group i, Yi represents the mean of group i, Y. . represents the overall mean of all groups, Si represents the sample variance of group i, N represents the total sample size, and k represents the number of groups.
Phylogenetic analysis
Phylogenetic analysis was conducted using single-copy gene families identified from the PIRATE results in the C. striatum core genome. Gene family sequences were retrieved from each genome and aligned using BLAST (default parameter). These sequences were then concatenated to create super-genes, which were used to build trees with the RAxML tools 49 using the GTRGAMMAI substitution model (-m PROTGAMMALGX -T 48 -N 100). The resulting phylogenetic tree was visualized with iTOL, 50 and the geographical locations and isolated tissues of each strain were also included.
Predictions of antibiotic resistance genes and virulence factors
To predict antibiotic resistance genes in C. striatum, we utilized the Comprehensive Antibiotic Resistance Database (CARD). 51 Firstly, the genomic data of C. striatum was translated into protein sequences, and then these sequences were compared to the CARD using Blastp. We only considered results that had an alignment coverage of ⩾80% and alignment identity of ⩾80% as antibiotic resistance genes.
For predicting virulence factors, we used Blastp to compare the protein sequences of C. striatum with the Virulence Factor Database (VFDB). 52 we only kept results with an alignment coverage of ⩾80% and alignment identity of ⩾60% as the virulence factors of it. To facilitate comparative analysis, the predicted genes were added to the phylogenetic tree.
Results
The pan-genome of C. striatum
After conducting genomic integrity analysis and average nucleotide identity analysis, we excluded 11 strains with an integrity level below 95% and 31 strains with an alignment coverage of less than 80%. The results of the BUSCO and average nucleotide identity analysis are shown in the Supplemental Figures 1 and 2.
The genome size of 314 strains ranged from 2.6 million base pairs to 3.6 million base pairs, with a GC percentage ranging from 57.2% to 59.5%. Subsequently, we proceeded with a pan-genomic analysis on these genomes, and the details of each strain are presented in Supplemental Table 1. CD-HIT clustering yielded 5692 gene families, with 32.4% (1845) present in the core genome, 41.5% (2362) in the accessory genome, and 26.1% (1485) in the unique genome (Figure 1). A detailed breakdown of each gene family is provided in Supplemental Table 2. Notably, the majority of gene families with a clustering threshold above 95% accounted for 83.2% and were mostly single-copy genes, with only a few fusion and fracture genes (Figure 1A), this finding suggest that C. striatum does not undergo large-scale rearrangement events. Figure 1B shows that the pan-genome size of C. striatum increases as more genomes are included in the analysis, indicating an open pan-genome for this species, this suggests that the species has the ability to acquire new genes in various environments by exchanging genetic material with other species through different mechanisms.

Pan-genomic characteristics of C. striatum: (A) multiple copies, division and fusion of gene families under each similarity threshold, (B) the pan-genomic size change model of C. striatum predicted by BPGP, and (C) KEGG annotation results of the core, accessory and singleton gene families of C. striatum pan-genome.
The gene families in the C. striatum pan-genome are predominantly associated with several KEGG pathways, namely Amino acid metabolism, Carbohydrate metabolism, Membrane transport, Replication and repair, and Signal transduction (Figure 1C). In the core genome, the most prevalent pathways include Amino acid metabolism, Carbohydrate metabolism, and Metabolism of cofactors and vitamins. On the other hand, the pathways that comprise accessory and unique genes, such as Cellular community, Circulatory system, Environmental adaptation, Immune system, Signaling molecules and interaction, and Substance dependence, may account for the phenotypic differences observed in C. striatum.
The intraspecific diversity of C. striatum
The results of the variance analysis conducted on the number of non-core genes and SNVs among different sources ofC. striatum showed significant differences (P ⩽ 6.2e−05), indicating that the genomic diversity and intraspecific diversity of the species were strong (Figure 2). The number of non-core genes and SNVs in the isolates from Beijing and Tangshan, China exhibited significant fluctuations, indicating strong intraspecific diversity in these 2 regions (Figure 2A and C). Similarly, the isolates from sputum, blood, and wound also showed a high degree of intraspecific diversity in terms of non-core genes and SNVs (Figure 2B and D). However, the number of non-core genes in strains isolated from skin was significantly lower than that in strains isolated from blood and sputum (Figure 2B), and the number of SNVs in these strains fluctuated less compared to strains isolated from other tissues (Figure 2D), indicating that the genome of strains isolated from skin was more conserved.
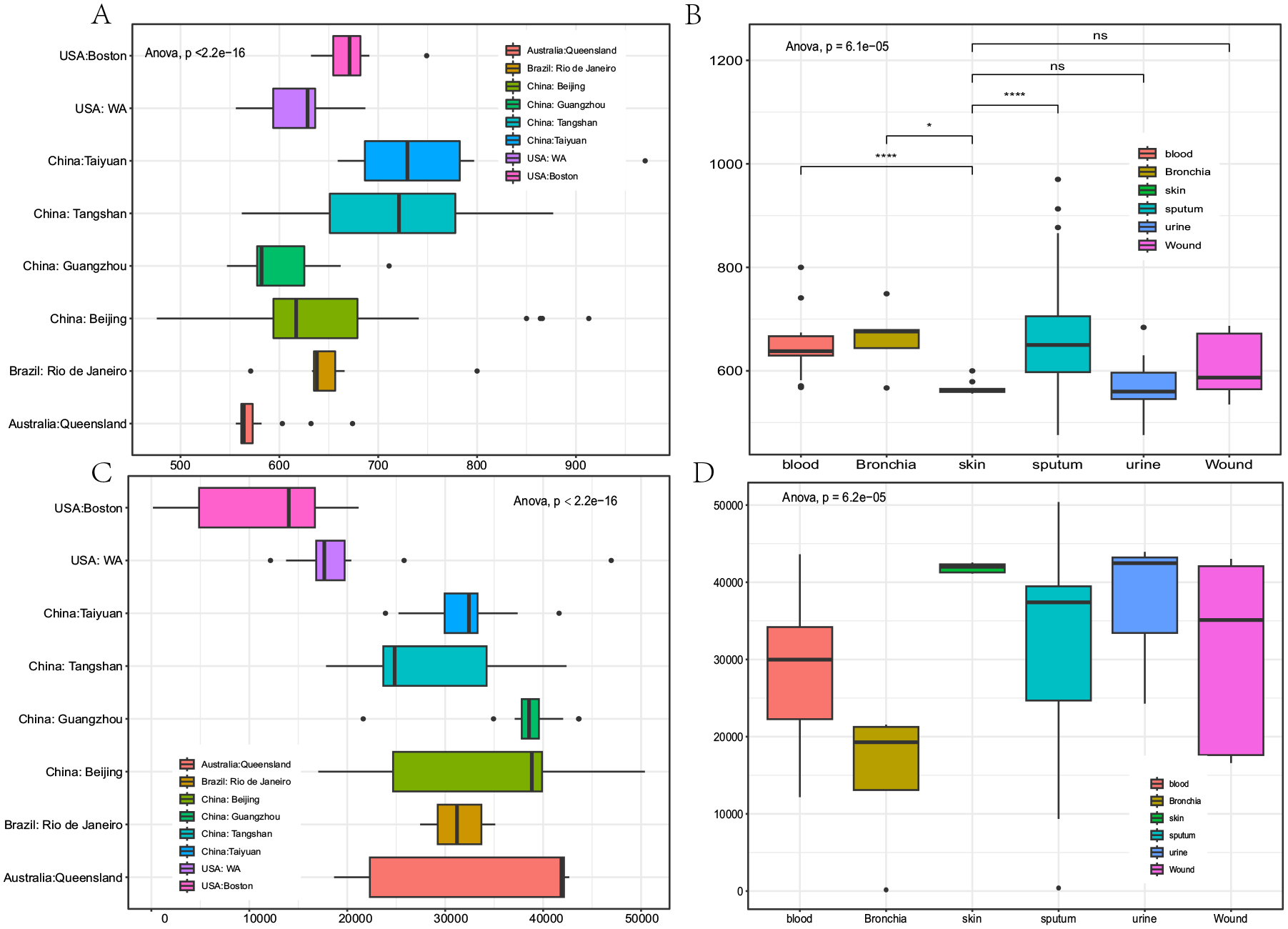
Differences in the number of non-core genes and genome-wide SNVs in C. striatum from different sources: (A and B) number difference and significance of non-core genes and (C and D) number differences and significance of genome-wide SNVs.
Phylogenetic analysis
The super-gene was created by concatenating 1273 single-copy genes from the core genome of C. striatum. To construct the phylogenetic tree, we utilized the maximum likelihood method with RAxML, and the tree also incorporated information on the isolation tissue and geographical location of the strains (Figure 3).
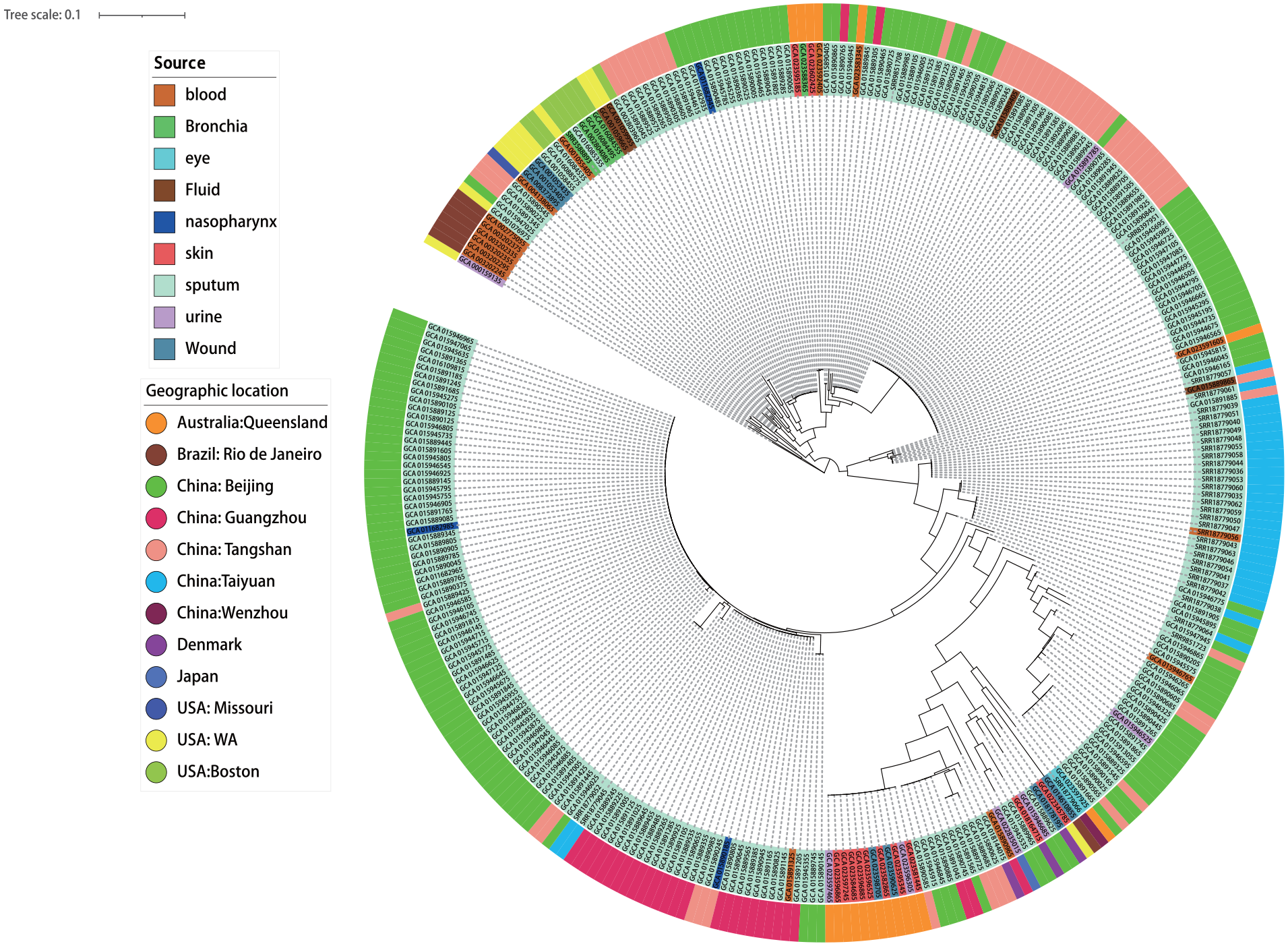
Phylogenetic tree of C. striatum.
The evolutionary tree displayed discrete distribution of strains from different sources, indicating a strong diversity. The strains isolated from Beijing and Tangshan, China showed a wide distribution in the phylogenetic tree, suggesting that they have been spreading for a long time, resulting in high intraspecific differences and strong diversity. In contrast, strains from Guangzhou and Taiyuan, China were more clustered in the tree. Strains isolated from sputum showed greater variability in the number of accessory genes and were distributed across various branches of the tree, indicating an open core and accessory genome. In contrast, strains isolated from skin tissue were more tightly clustered in the tree, and showed less variation in the number of non-core genes and SNVs, suggesting a more conserved core and accessory genome.
Prediction of resistance genes
Aligned prediction led to the identification of 53 resistance genes in the C. striatum genomes, which are presented in detail in Supplemental Table 3. We compared resistance genes within the same gene family using a phylogenetic tree.
Among the strains, 77.7% contained 2 or more resistant genes (Figure 4). The ermX gene, encoding an rRNA methyltransferase that protects ribosomes from inactivation by antibiotics such as macrolides, lincosamides, and streptogramins, was present in 90% of the strains.53,54 The tetW gene, which encodes a ribosome protective protein that confers resistance to tetracycline, was found in 61% of the strains.55,56

Distribution of the antimicrobial resistance genes in C. striatum.
The sul1 gene, a sulfonamide-resistant dihydropteroate synthase of Gram-negative bacteria, was detected in 58% of the strains and was linked to other resistance genes of class 1 integrons. 57 The qacE gene, present in 57% of the strains, confers resistance to antimicrobials. 58
Aminoglycoside N-acetyltransferase-encoding genes AAC(6) and AAC(6′) -Ib-cr were identified in 57% of strains, and were responsible for inactivating aminoglycoside antibiotics by acetylating the 6-amino group of the compound. 59 The APH (6′) and APH (3′) genes, encoding aminoglycoside O-phosphotransferases, were detected in 32% of strains, and inactivate antibiotics, especially streptomycin. 60 The Cstr_tetA (tetAB) gene was identified in 30.2% of strains, is located in the plasmid of C. striatum, encodes the ABC transporter, and confers resistance to tetracycline and oxytetracycline. 15
The ANT (3") gene family was detected in 38.9% of strains, and it encodes a class of aminoglycoside O-nucleotidyltransferases with modified region specificity based on the 3″-hydroxyl group of the respective antibiotic. These enzymes inactivate aminoglycoside antibiotics by transferring the AMP group from the ATP substrate to the 3″-hydroxyl group of the compound. 61
Most strains from different tissues harbored more than 2 antimicrobial resistance genes. However, in a specific region of the phylogenetic tree, most strains only carried the ermX gene. These strains were mainly isolated from sputum, skin, and wound and were closely related to skin strains. This observation suggests that C. striatum in skin may be exposed to less antibiotic selection pressure and may carry fewer resistance genes. There was no significant difference in the distribution of antimicrobial resistance genes among strains from different geographical locations.
Prediction of virulence factors
In our study, we identified 42 virulence factors in C. striatum genomes and associated 15 genes with these factors, as detailed in Supplemental Table 4. Among these, the sigA and sodA genes were found in all strains (Figure 5). SigA, also known as rpoV in Mycobacterium tuberculosis, is thought to direct extracellular function and various other stress responses, such as temperature, oxidative stress, pH, and macrophage infection, 62 it is thought to direct extracellular function and various other stress responses (temperature, oxidative stress, pH, and macrophage infection) . On the other hand, the sodA gene encodes an iron-dependent enzyme that is crucial for the survival of pathogens within the cell during infection. 63

Distribution of virulence genes in C. striatum.
We also found that the SpaD and SpaE genes were present in 98% of the strains. In Corynebacterium diphtheriae, these 2 genes have been reported to be involved in the formation of pili, which enables the strains to adhere specifically to human pharyngeal epithelial cells. 64 Additionally, the Srtc and SrtB genes were identified in 48.7% of strains. Srtc is a fimbrial-associated sortase, and in Corynebacterium diphtheriae, pili are produced by the sortase mechanism. 65 The SrtB gene encodes collagen-binding protein in Clostridium difficile, which binds to human complement C1q, and thus may be involved in host immune escape mechanisms and is important in early biofilm formation.66 -68
Furthermore, The fagABCD operon genes were identified in 14.9% of the strains, with 4 genes present in each strain, and these 4 genes are related to iron acquisition. 69 The regX3 gene, which encodes a sensory transduction protein, was identified in 89.8% of the strains. 70 Interestingly, isolates from different geographical locations and tissues did not show significant differences in virulence factors.
In summary, our findings shed light on the virulence factors of C. striatum and provide important insights into the pathogenicity of this bacterium. Further studies are needed to elucidate the mechanisms underlying the roles of these virulence factors in C. striatum infections.
Discussion
In this study, we performed a pan-genomic analysis of 314 genomes of C. striatum, revealing an open pan-genome composed of 5692 gene families. This result is consistent with the findings of Jesus et al, 28 but our study used a larger number of strains and identified more gene families. Through KEGG pathway annotation of gene families, we observed that the majority of the genes were associated with transcription and metabolism, while accessory genes and unique genes composed pathways such as Cellular community, Circulatory system, Environmental adaptation, and Immune system. Notably, we identified novel unique genes related to infectious diseases, biodegradation, and metabolism.
We conducted an analysis of variance (ANOVA) on the number of non-core genes and genome-wide SNVs in C. striatum obtained from various sources. The results revealed significant differences among the sources, providing evidence for the presence of intra-species diversity within C. striatum. We observed the greatest variation in strains from Beijing, China. This finding suggests that prolonged transmission events in this region may have resulted in large intra-species differences. This hypothesis is supported by a previous study showing that C. striatum was transmitted for 2 years in a hospital in this region. 36 The strains isolated from skin had significantly fewer accessory genes and unique genes than other tissues, and were more concentrated in the phylogenetic tree. Additionally, these strains had fewer resistance genes compared to other tissues. Our analysis suggests that C. striatum on the skin, as a normal microbe, 71 experiences less selective pressure such as antibiotics and has fewer opportunities for gene exchange with the environment, resulting in a relatively conserved genome. However, strains from other tissues, such as sputum, wound, and blood, are subject to greater selection pressure, resulting in an open genome with large intra-specific differences. This phenomenon has not been previously reported.
We identified 53 antimicrobial resistance genes, which were grouped into a total of 11 gene families. Most of these genes were located in mobile genetic elements. The ermX, cmx, strA, and strB, are all located on the pTP10 plasmid of C. striatum. 15 The pTP10 plasmid is comprised of 8 regions, consisting of 2 regulatory regions and 6 transport regions for resistance genes. 12 The ermX gene is situated on transposon Tn5432 within the first transport region, which contains 2 identical insertion sequences on either side (IS1249). 15 Tn5432 harbors the ermX gene, which confers resistance to lincomycin and macrolide antibiotics, including clindamycin and erythromycin. 54 Moreover, the cmx, strA, and strB genes are carried by Tn5717 and Tn5716 on the pTP10 plasmid, respectively. The cmx gene confers resistance to chloramphenicol, while the strA and strB genes confer resistance to aminoglycoside antibiotics, such as streptomycin. 15
Previous studies have reported the presence of the tetW gene, associated with resistance to tetracycline. This gene has been found to be carried by IS3504, IS3503, and IS3502, which are insertion sequences identified within the genome of Corynebacterium jeikeium.13,35 Additionally, sul1, AAC(6′), AAC(6′)-Ib-cr and qacE genes have been reported to be present on class 1 integron in C. striatum. 28 This integron possess the capability of being inserted, removed, rearranged, and expressed through site-specific recombination systems, making them effective vectors for the transfer of genetic material within and between species. 72 It is worth noting that the presence of class 1 integron in Pseudomonas aeruginosa has been linked to the development of multidrug-resistant phenotypes, 72 further emphasizing their significance in promoting antibiotic resistance. However, the positions of less-studied genes, such as APH(6), APH(3′), and ANT(3"), within the C. striatum genome have not been established. In contrast to the study by Jesus et al, 28 our research used a larger dataset, which allowed us to identify additional genes, including the new gene family ANT(3″). This family encodes aminoglycoside O-nucleotide transferases that transfer the AMP group from the ATP substrate to the 3″-hydroxyl group of the compound, resulting in the inactivation of aminoglycoside antibiotics. 61
We detected a total of 42 virulence factors, which were associated with 15 genes, with fagABCD operon mainly related to iron uptake, also present in Corynebacterium pseudotuberculosis.73,74 The SpaD, SpaE, Strb, and SrtC genes play key roles in the formation of fimbriae and biofilms, which contribute to the enhanced colonization ability of C. striatum.67,75 Previous studies have demonstrated that C. striatum has the capability to adhere to both hydrophilic and hydrophobic abiotic surfaces, forming mature biofilms specifically on polyurethane and silica catheter surfaces.20,21 These findings align with the conclusions drawn from our study. Nonetheless, we were able to identify 4 new virulence genes, including sodA, groEL2, sigH, and regX3. The sodA gene was present in all strains, encoding an iron-dependent enzyme crucial for the survival of pathogens within host cells during infection. 63 The SigH gene, found in 7.6% of the strains, exhibited similarity to the sigma R of Streptomyces species and was implicated in response to heat shock and oxidative stress.76 -78 On the other hand, the groEL2 and regX3 genes have been poorly studied, and their function remains unknown.
Conclusions
Our study revealed that C. striatum exhibits strong intraspecific diversity and an open pan-genome, with new genes acquired through gene exchange events under different selective pressures. Specifically, the skin tissue isolates had a relatively conserved accessory genome and core genome, with a lower number of drug-resistant genes compared to other tissue isolates, likely due to the lower selective pressure on the skin tissue. Nevertheless, drug resistance genes and virulence factors were widely distributed in the population, with 77.7% of the strains carrying 2 or more drug resistance genes, most of which were located on mobile genetic elements and mainly related to resistance against aminoglycosides, macrolides, and tetracycline. Virulence factors were mainly related to pathogenic bacterial survival within the host, iron uptake, pili, and early biofilm formation. Our study is the first to examine the genetic characteristics of this emerging multidrug-resistant clinical pathogen population, confirming the potential of C. striatum to become a clinical pathogen at the genetic level. This finding addresses the current knowledge gap in the genetic characteristics of C. striatum population and provides new insights into the genetic characteristics of this emerging multidrug-resistant clinical pathogen population, which could inform future research and clinical practice in the diagnosis, prevention, and treatment of related diseases.
Supplemental Material
sj-docx-1-evb-10.1177_11769343231191481 – Supplemental material for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen
Supplemental material, sj-docx-1-evb-10.1177_11769343231191481 for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen by Junhui Qiu, Yulan Shi, Fei Zhao, Yi Xu, Hui Xu, Yan Dai and Yi Cao in Evolutionary Bioinformatics
Supplemental Material
sj-xls-2-evb-10.1177_11769343231191481 – Supplemental material for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen
Supplemental material, sj-xls-2-evb-10.1177_11769343231191481 for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen by Junhui Qiu, Yulan Shi, Fei Zhao, Yi Xu, Hui Xu, Yan Dai and Yi Cao in Evolutionary Bioinformatics
Supplemental Material
sj-xls-3-evb-10.1177_11769343231191481 – Supplemental material for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen
Supplemental material, sj-xls-3-evb-10.1177_11769343231191481 for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen by Junhui Qiu, Yulan Shi, Fei Zhao, Yi Xu, Hui Xu, Yan Dai and Yi Cao in Evolutionary Bioinformatics
Supplemental Material
sj-xls-4-evb-10.1177_11769343231191481 – Supplemental material for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen
Supplemental material, sj-xls-4-evb-10.1177_11769343231191481 for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen by Junhui Qiu, Yulan Shi, Fei Zhao, Yi Xu, Hui Xu, Yan Dai and Yi Cao in Evolutionary Bioinformatics
Supplemental Material
sj-xls-5-evb-10.1177_11769343231191481 – Supplemental material for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen
Supplemental material, sj-xls-5-evb-10.1177_11769343231191481 for The Pan-Genomic Analysis of Corynebacterium striatum Revealed its Genetic Characteristics as an Emerging Multidrug-Resistant Pathogen by Junhui Qiu, Yulan Shi, Fei Zhao, Yi Xu, Hui Xu, Yan Dai and Yi Cao in Evolutionary Bioinformatics
Footnotes
Acknowledgements
We acknowledged Yi Cao of Sichuan Provincial Key Laboratory of Microbiology and Metabolic Engineering and Yan Dai of Wound treatment center of West China Hospital of Sichuan University for their guidance on this study. The authors acknowledge the anonymous reviewers for their valuable suggestions that helped improve the quality of the manuscript.
Author Contributions
Yi Cao and Yan Dai conceived the idea. Yi Cao and Hui Xu supervised the study. Junhui Qiu, Yulan Shi, Fei Zhao and Yi Xu analyzed and interpreted the data. Junhui Qiu drafted the manuscript. Yi Cao and Yan Dai revised the manuscript critically. All authors read and approved the manuscript.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Natural Science Foundation of China (32271535, 32071479); Department of Science and Technology of Sichuan Province (2021YJ0024, 2022NSFSC0243, 2022NSFSC0119); Special fund for West China nursing discipline development of Sichuan University (HXHL19006).
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.